First steps in R
Bioinformatics and Biostatistics Hub, Computational Biology Department

1 Get ready for the course
Before the course starts, you should have installed R and RStudio on your computer (see below). RStudio is highly recommended, especially if you are a beginner.
1.1 About R
This course will use a programming language oriented towards data analysis and statistics: R.
It can be useful for:
- Publication ready graphs
- Biological analysis libraries
- Statistical testing
- Many more: table reformatting, maps, interactive visualizations, etc…
R can be used in an interactive console: typing and executing commands one by one.
Another way to work with R is to prepare a list of commands in a text file (a script, which is a kind of program) and then tell R to execute them. This is an excellent way to have reproducible and well documented analyses.
1.2 About RStudio
RStudio is an application in which you can load data, execute R commands, prepare scripts, get information on commands and environments (the current state of your work), visualize graphics and more.
You do not need RStudio to work with R, but it will make your life easier during this course.
1.3 Installing R
R can be obtained from the following site: https://cran.rstudio.com/
Try to install version 3.5 or above (and preferably at least 3.6). If your operating system provides R already, but in a slightly earlier version, most things in the present course will probably work fine, but this might not be true for later R-based courses, and you will run into trouble sooner or later.
For those using a Debian or Ubuntu based Linux, we suggest reading the following material before proceeding to install R: https://github.com/blaiseli/R_install_notes/blob/master/Install_Linux.md. Some of the content may also be helpful for users of other types of Linux distributions.
Windows users should refer to the following page: https://cran.rstudio.com/bin/windows/base/
Mac OSX users should refer to this one: https://cran.rstudio.com/bin/macosx/
1.4 Installing RStudio
You can obtain RStudio from this page: https://www.rstudio.com/products/rstudio/download/
Choose the free version (“RStudio Desktop, Open Source Licence”).
Download and run the “installer” suitable for your operating system.
1.5 Course material
- The most up-to-date version of the present course support should be available at http://hub-courses.pages.pasteur.fr/R_pasteur_phd/First_steps_RStudio.html
- If you spot typos, errors, ambiguities, etc., you can sign in on our local gitlab server using your Pasteur ID (if you are a member of the Institut Pasteur) or using a github account to submit an issue here: https://gitlab.pasteur.fr/hub-courses/R_pasteur_phd/issues
- You can get the full course material (html page, data for the exercises, and more) in the following zipped archive (recommended): http://hub-courses.pages.pasteur.fr/R_pasteur_phd/2020-03/First_steps_RStudio.zip
The course contains small exercises (indicated with  ) to help you memorize the techniques we present. Try to search by yourself, then discuss with your neighbours (if you want), before clicking on the “Hint” buttons. There are often several ways to solve a problem when programming. If you come up with solutions other than the ones we propose, while still using techniques already seen in the course, don’t hesitate to share them with us.
) to help you memorize the techniques we present. Try to search by yourself, then discuss with your neighbours (if you want), before clicking on the “Hint” buttons. There are often several ways to solve a problem when programming. If you come up with solutions other than the ones we propose, while still using techniques already seen in the course, don’t hesitate to share them with us.
The buttons with the  icon open sections that will not be treated during the course. Feel free to study them when you have free time (if the course pace is too slow for you, for instance).
icon open sections that will not be treated during the course. Feel free to study them when you have free time (if the course pace is too slow for you, for instance).
Here’s one already!
 ) button.
) button.The material for the other modules of the course is (or will soon be) available at https://c3bi.pasteur.fr/bioinformatics-program-for-phd-students-2019-2020/.
2 Smooth introduction to R
2.1 The RStudio interface
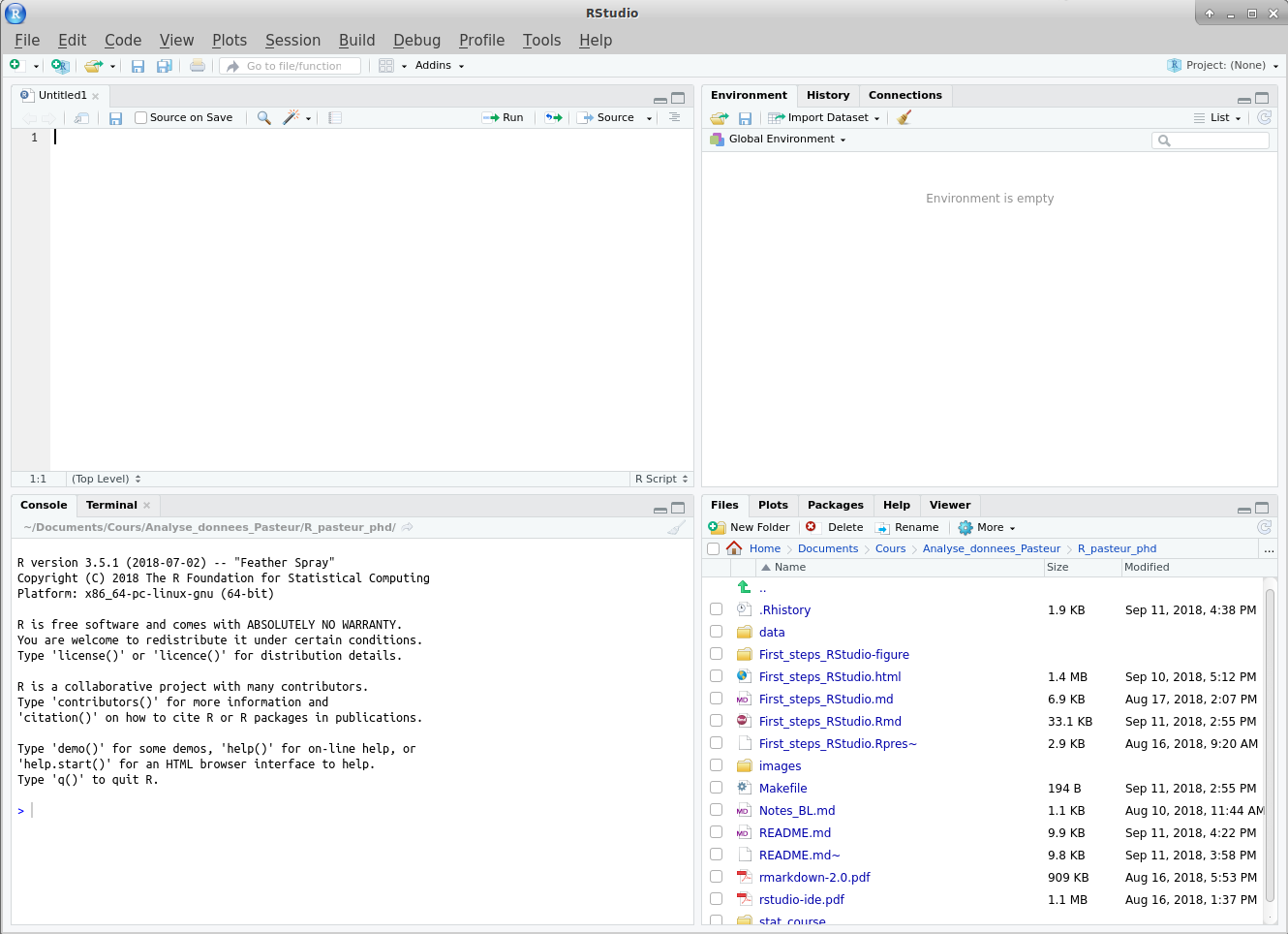
By default, RStudio opens with three or four panes, each containing several tabs:
- The top-left pane (if present, otherwise click
 ) is the editor, where you can write scripts, and other types of documents. This is also where data frames can be visualized.
) is the editor, where you can write scripts, and other types of documents. This is also where data frames can be visualized. - The bottom-left one contains the “Console”, where you can directly type R commands.
- The top-right one contains information about the “Environment”: values currently defined in your R session.
- The bottom-right one contains various tabs, including the following ones:
- a file explorer
- a help viewer
- a plot visualizer
- a package manager (packages are a way to extend the functionalities of R).
2.2 Loading data in RStudio
A frequent use of R is to analyse data stored as tables in files.
2.2.1 Quick dive in Milieu Intérieur dataset
The “Milieu Intérieur” project is an ambitious population-based study coordinated by the Institut Pasteur, Paris. The objective is to dissect the interplay between genetics and environment and their impact on the immune system (https://research.pasteur.fr/en/program_project/milieu-interieur-labex/).
Download the Milieu Intérieur dataset from http://hub-courses.pages.pasteur.fr/R_pasteur_phd/data/mi.csv, and remember where in your computer it was saved.
The “Environment” tab of the top-right pane of RStudio contains an “Import Dataset” button ( ) that can be used to load various kind of files.
) that can be used to load various kind of files.
Open the “Import Dataset” menu and choose “From Text (base)…”.
A file browser will open. Select the file you just downloaded.
You should see a window presenting you options related to how the content of the file should be interpreted, similar to the following:
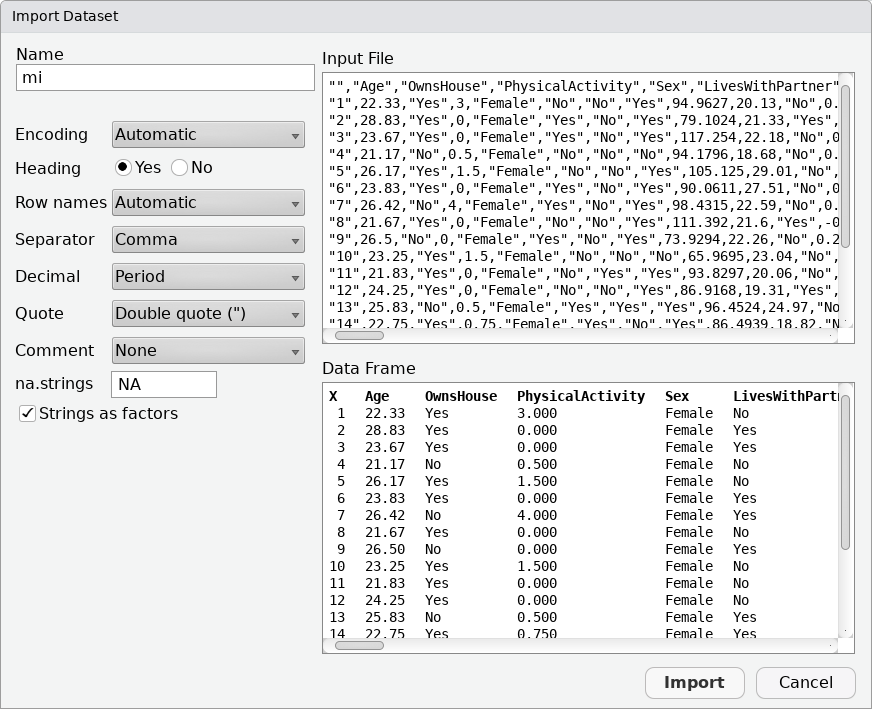
Most default values should be OK. Be sure that the “Heading” option is set to “Yes” and select “Use first column” for the “Row names” option, then click on the “Import” button.
A tab should open in the top-left pane displaying the content of the file. These data represent information pertaining to individuals that participated to the “Milieu Intérieur” study. Lines correspond to individuals (“observations”), and columns to various pieces of information obtained for them (“variables”).
In the “Environment” tab of the top-right pane of RStudio, you have access to more information about this table:
- next to the name of the table (
mi, if you used the default choice at import), the number of observations and variables in the table; - below (if you click on the arrow next to the name of the table), a listing describing how the data for each column is represented in R.
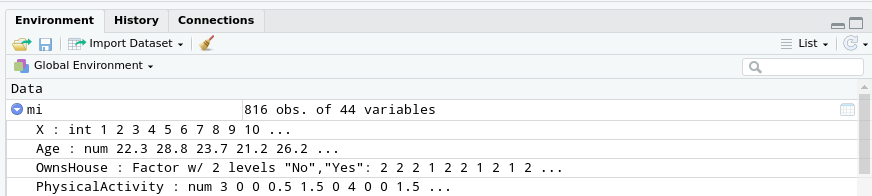
You can see that the information consists in numerical and textual information.
What we obtained by loading this file is called a “data frame” in R. A lot of things can be done using data frames, and we’ll learn some of it later in this course.
Briefly, and to wet your appetite, a specific column of a data frame can be accessed by using the name of the data frame (here mi), a dollar sign $ and the name of the desired column (here for example Age):
mi$Age [1] 22.33 28.83 23.67 21.17 26.17 23.83 26.42 21.67 26.50 23.25
[ reached getOption("max.print") -- omitted 806 entries ]You want to visualize the age distribution of this dataset ? To plot a histogram, just use the hist function:
hist(mi$Age)
A function is a predefined set of computations that can be re-executed, possibly with variations, depending on its arguments (the things given between the parentheses). Pre-defined functions, like hist help make R very useful for non-programmers.
But we are getting ahead of ourselves and we will see more on functions, data frames manipulation and data visualization later on\(\dots\) Before being able to actually analyse data, we will have to learn about the various ways used to represent data in R, and what kind of things can be done with data on a basic level.
2.3 R as a calculator
In the “Console” part of RStudio, you can see a prompt: This > symbol invites you to type commands.
Try the following (validate using the “return” key after typing):
> 1 + 1[1] 2It seems R gave the correct result (ignore the [1] for now).
Note that some complicated calculations may take a non-negligible time. A  button should be visible until the calculation ends. If you suspect something is going wrong, use it to interrupt the calculation.
button should be visible until the calculation ends. If you suspect something is going wrong, use it to interrupt the calculation.
Once R is ready to take new commands, you should see a prompt (>) again in the console.
You can of course also subtract (-), multiply (*) and divide (/). Feel free to experiment with the usual mathematical operators.
2.4 Numbers
Divisions may give answers with a dot as decimal separator:
> 2 / 3[1] 0.6666667 The result can be rounded for the display. The actual value may also sometimes not be mathematically exact. This is due to limitations inherent to the way numbers are encoded by the computer.
The result can be rounded for the display. The actual value may also sometimes not be mathematically exact. This is due to limitations inherent to the way numbers are encoded by the computer.
Use a dot, not a comma when writing numbers with decimals (called double in R):
> 3.14[1] 3.14By the way, R knows about \(\pi\):
> pi[1] 3.141593Scientific notation is allowed, and could be useful:
> 5e-3[1] 0.0052.5 Variables
2.5.1 Defining and using variables
Like the predefined pi, you can temporarily “save” values in the computer memory by associating them to a name:
> my_variable <- 2 + 2Some people tend to use the equal sign (=) instead of the above left-pointing arrow (<-). Both work in R.
You can then use the corresponding value by simply typing the associated name:
> my_variable[1] 4> my_variable * 5[1] 20This is called a variable: the associated value can be updated by later commands:
> my_variable <- 5
> my_variable[1] 5> my_variable <- 3
> my_variable[1] 3> my_variable <- 2 * 3
> my_variable[1] 6 By the way, you can re-use the previous commands in the console by navigating the “command history” using the up and down arrows of your keyboard. You can then modify the selected command if needed (use the left and right arrows to get to the part you want to edit).
By the way, you can re-use the previous commands in the console by navigating the “command history” using the up and down arrows of your keyboard. You can then modify the selected command if needed (use the left and right arrows to get to the part you want to edit).
Note that the older values are now lost (variables are also lost when you quit or restart R).
You will use variables a lot. You can use them to store many different types of values that we will see later. For instance, the “Milieu Intérieur” data frame we previously loaded from a file is stored using a variable named mi.
Note that this notion of “variable” (in the “computer programming” sense) is not exactly the same as the one we used when referring to columns in the data frame, which is more a notion belonging to the data science field.
You can even store functions (sequences of commands that can be re-used).
You can use a variable as many times as you wish in a calculation, and you can even use it to create another variable:
> radius <- 4
> area <- pi * radius * radius
> area[1] 50.26548 Another handy feature provided by RStudio is code completion. You will notice it after typing three characters: a pop-up will appear giving you the variables and functions starting with these characters and available in the current environment. This pop-up is giving you a brief description as well. You can make the code completion pop-up also appear by pressing the TAB key (the one with a kind of arrow, located at the left of the first row of letters of most computer keyboards).
You can also see the variables you defined and their current value in the “Environment” tab (top-right pane).
2.5.2 Variable naming rules
Variable names need to conform to certain rules. You should be relatively safe if you stick to the following:
- Use only:
- plain ASCII letters (“A” to “Z” and “a” to “z”);
- numbers;
- underscore ("_").
- Don’t start a variable name with underscore or a number.
- Avoid single letter unless meaning is obvious.
- Avoid using names that already exist (that’s the tricky part).
This is OK:
> five_point_6 <- 5.6This is not (try it, you will get an error):
> 6_times_4 <- 24## Error: <text>:1:2: unexpected input
1: 6_
^You can do this, but you shouldn’t:
> pi <- 3.14If you did it nonetheless (you fool…) the older and more accurate value of pi is still available as base::pi.
The names of your variables should help you and others understand what the code is supposed to do.
You will sometimes see dots (.) in variable names. This is allowed in R, but in many programming languages, dots have special meanings. You’d better avoid taking the habit of using dots in variable names. This will make the transition less painful if you ever decide to learn other languages.
2.6 Intermission: Scripts in RStudio
2.6.1 Getting organized
Above the console, there is a space where you can take note of the commands you want to remember, and save this to a (plain text) file.
This is actually intended to create a script. A script is a file containing a succession of commands that can be executed to do all the corresponding computations. Just put a # before the parts that are not valid R commands.
This is called “commenting”, and it is a good practice to put such comments in your scripts, especially in parts that took you time to get right. That helps you remember what your intentions were when you wrote the commands (and can also help others, if you share your scripts).
(You can use the button to create a new script or in the menu “File” > “New File” > “R Script”.)
Small script example:
2.6.2 Executing saved scripts
 Execute code on the current line (or block of selected lines) in the console below (shortcut: (Ctrl or Cmd) + Enter).
Execute code on the current line (or block of selected lines) in the console below (shortcut: (Ctrl or Cmd) + Enter). Execute the whole script in the console below (shortcut: (Ctrl or Cmd) + Shift + Enter).
Execute the whole script in the console below (shortcut: (Ctrl or Cmd) + Shift + Enter).
(You can also execute an R script outside RStudio, using the Rscript command from a command line interface.)
Example of execution of the current line:
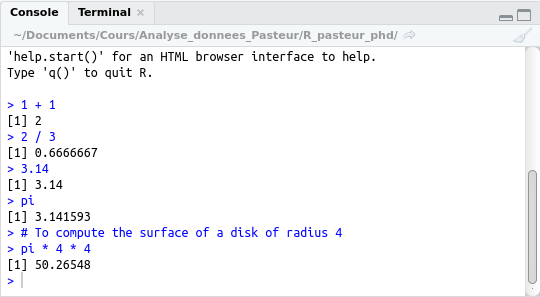
(The preceding comments have also been “executed”: they appear in the console, but have no other effects.)
2.7 Truth values (“Booleans”)
You can compare numbers using comparison operators:
3 * 2 > 4[1] TRUE1 <= -3[1] FALSEDouble equal == is used to test equality:
5 == 2 + 3[1] TRUE!= (not equal) is used to test inequality:
1 + 3 != 4[1] FALSEThe result of a comparison is a value that tells you whether what you wrote is true or not. Such values are called Boolean values, or “Booleans” (after the logician George Boole).
They must be written in capitals:
TRUE[1] TRUEFALSE[1] FALSEOtherwise, R looks for a variable with that name:
true## Error: object 'true' not found Don’t name your variables with names such as true or false, you might get bitten.
Code failing with an error message is often better than undetected mistakes.
2.7.1 Boolean operations
You can use Booleans to compute more Booleans, using logical operators:
TRUE & FALSE # And[1] FALSETRUE | FALSE # Or[1] TRUE!FALSE # Not[1] TRUEFALSE & ((1 == 1) | TRUE)[1] FALSEIn the last command, we mix comparison expressions (that have Boolean results) with “literal” Booleans (FALSE and TRUE) The whole result has a Boolean value.
Expressions can be composed of a hierarchy of expressions combined with operators. Operators have precedence rules, telling what operations should be evaluated first.
To be sure that expressions will be evaluated in the intended order, we can use parentheses in complex expressions, as above. Here is a decomposition of the evaluation:
1 == 1is TRUE, therefore((1 == 1) | TRUE)isTRUE.- This expression is hence equivalent to
FALSE & TRUE, which isFALSE.
(Parentheses can be used in any expression, not just in Boolean calculations.)
2.8 Strings (text)
Computer programs are much more useful for humans if they can communicate with them using text, and let them manipulate text, and not just simple mathematical objects such as numbers or Booleans. Indeed, text is a powerful tool to convey complex information. Text can be used to encode information in various ways.
Fortunately, R offers (and heavily uses) the possibility to represent text.
We can define character strings (or just “strings”) variables to represent text, placing it "between quotes":
selected_color <- "blue"
file_name <- "counts.csv" Note that there are no quotes when one writes TRUE, FALSE or a variable name. This is a very important difference to keep in mind.
We will see later what can be done with strings.
3 Vectors
3.1 Creating or getting vectors
You can group values of the same type in a vector, using c (combine).
Numbers:
my_numbers <- c(6, -1, 0, 6)
my_numbers[1] 6 -1 0 6Strings:
phenotype <- c("round", "wrinkled", "round", "wrinkled")
phenotype[1] "round" "wrinkled" "round" "wrinkled"Booleans:
tests <- c(TRUE, FALSE, FALSE, TRUE)
tests[1] TRUE FALSE FALSE TRUEOf course, values in a vector can be the result of a computation:
c(FALSE, 1 == 1, 2 != 2)[1] FALSE TRUE FALSEThey can be variables too:
a <- 2
b <- 3 + 4
c(a, b)[1] 2 73.1.1 Named vectors
There is a potentially nice feature in R that string-capable people like you can benefit from: named vectors.
Elements in a vector can indeed have names.
To have a named vector, you can give names to its elements at creation time, by prefixing each element with the desired name and an equal sign:
mix <- c(buffer=2.5, MgCl2=0.75, dNTP=0.5, primer1=0.5, primer2=0.5, water=20.125, Taq=0.125)The names are then displayed above the corresponding values when you display the vector:
mix buffer MgCl2 dNTP primer1 primer2 water Taq
2.500 0.750 0.500 0.500 0.500 20.125 0.125 3.1.2 Automatic conversions
Be careful. Vectors can only contain values of the same nature. If you try to create a vector of values of mixed types, R will automatically convert values in a way that may or may not be what you want. You will not get warnings.
c(1, "two", 3, "four")[1] "1" "two" "3" "four"c(TRUE, "OK", FALSE, "Ouch!")[1] "TRUE" "OK" "FALSE" "Ouch!"c(1, FALSE, "three")[1] "1" "FALSE" "three"In the above cases, there is no reasonable way arbitrary words can be interpreted as numbers or Booleans. The only option left is to turn everything into strings (just add quotes!).
Guess what happens when you just combine numbers and Booleans?
3.1.3 Sequences of numbers
There’s an useful shortcut to create a vector of successive numbers:
2:7[1] 2 3 4 5 6 7a:b[1] 2 3 4 5 6 7-4:4[1] -4 -3 -2 -1 0 1 2 3 4You can even generate decreasing series:
0:-5[1] 0 -1 -2 -3 -4 -53.1.4 Extracting columns from a data frame
If you remember well, we saw how to get a column from our mi data frame using the $ sign (mi$Age, for instance). Data frame columns are actually vectors. Try to save the ages from that data frame in an ages variable.
3.2 Vectorized operations
Certain operations can be applied on all the values in a vector simultaneously:
1:5 + 2[1] 3 4 5 6 7!c(FALSE, TRUE, TRUE, FALSE)[1] TRUE FALSE FALSE TRUEc(6, 4, 2, 1, 3, 5) < 4[1] FALSE FALSE TRUE TRUE TRUE FALSEYou can use this to easily compute the quantities of reagents when preparing a mix in the lab, if you have the recipe for a certain amount of mix (as stored in the mix variable previously) and want another amount:
original_mix_amount <- 25
desired_mix_amount <- 20
mix * desired_mix_amount / original_mix_amount buffer MgCl2 dNTP primer1 primer2 water Taq
2.0 0.6 0.4 0.4 0.4 16.1 0.1 Easy parallelized cross-multiplication!
These vectorized operations are very powerful, especially when dealing with large amounts of data. They are widely used by R practitioners, and you will use them during this course.
3.3 Operations between vectors
It is also possible to apply operations between two vectors.
c(1, 2, 3, 4) + c(2, 4, 6, 8)[1] 3 6 9 121:3 * 1:3 # equivalently: (1:3) ^ 2[1] 1 4 9The result is a vector in which the values are obtained by applying the operation on elements at the corresponding positions in the original vectors.
3.3.1 Value recycling
Now try to combine vectors of different sizes. Does it work? If yes, what happens?
Suppose we have the following vectors:
v1 <- 1:6
v2 <- 1:3 Without entering the computation in R, think what would be the result of v1 <= v2?
3.4 Combining vectors
You can add one or more values to a vector using c:
euro_bikes <- c("Pinarello", "BMC")
amer_bikes <- c("Cannondale", "Kona", "Specialized")
bikes <- c(euro_bikes, amer_bikes)
bikes[1] "Pinarello" "BMC" "Cannondale" "Kona" "Specialized"(This operation is sometimes called “concatenation”.)
It is also possible to prepend or append just one value to a vector:
c("Peugeot", bikes)[1] "Peugeot" "Pinarello" "BMC" "Cannondale" "Kona"
[6] "Specialized"c(bikes, "Raleigh")[1] "Pinarello" "BMC" "Cannondale" "Kona" "Specialized"
[6] "Raleigh" 3.5 Indices and length
Ask R to display a looong vector:
21:60 [1] 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45
[26] 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60The numbers between square brackets are here to help us count the number of elements. These are indices of the elements beginning a displayed line: 21 is the 1\(^{st}\) element, 44 is the 24\(^{th}\).
One can get the number of elements in a vector using the predefined length function:
length(21:60)[1] 40Functions are executed by writing their names followed by opening and closing parentheses. Between parentheses are the arguments of the function, if any. Here, we give a vector to the length function, and it gives back a result, which is the number of elements in the vector.
We’ll see more functions later. We have actually already seen another one: c is a function that can have more than 1 argument.
And another very useful function is sum:
sum(c(1, 1, 1))[1] 3 Given the following vector, can you think of a way to compute the number of elements above 5?
nums <- c(3, 7, 2, 3, 2, 8, 6, 2, 6)3.6 Accessing vector elements
3.6.1 Access by index
One can take elements in a vector using square brackets and their indices:
ages_toy <- c(34, 3, 18, 5, 56, 41, 23, 101)
ages_toy[3] # one element[1] 18ages_toy[3:6] # a slice[1] 18 5 56 41ages_toy[c(8, 6, 4, 2)] # elements in arbitrary order[1] 101 41 5 3 How would you get the last element?
One can also change the value of an element:
ages_toy[1] 34 3 18 5 56 41 23 101# It's the little child's birthday!
ages_toy[2] <- ages_toy[2] + 1
ages_toy[1] 34 4 18 5 56 41 23 101(Notice how the second element has been incremented.)
You will encounter other similar situations in R, where the same syntax can be use either to get or set a value.
We previously constructed a vector containing two European bicycle brands first, then three US brands.
bikes[1] "Pinarello" "BMC" "Cannondale" "Kona" "Specialized"Try to insert the Taiwanese brand "Giant" between the two parts.
3.6.2 Boolean sub-setting
A vector of Booleans can be used to extract portions of another vector:
ages_toy[1] 34 4 18 5 56 41 23 101ages_toy[c(
TRUE, FALSE, FALSE, FALSE,
TRUE, TRUE, FALSE, TRUE)][1] 34 56 41 101Only those values at positions where the vector of Booleans contains TRUE are selected.
This can be very useful, because a vector of Booleans can be obtained by applying to a vector an operation that results in a Boolean:
is_old <- ages_toy > 30
is_old[1] TRUE FALSE FALSE FALSE TRUE TRUE FALSE TRUEYou can therefore easily select the ages of the old guys in one command:
ages_toy[is_old][1] 34 56 41 101You can write this directly in one step:
ages_toy[ages_toy > 30][1] 34 56 41 101An interesting function is which, that gives the indices of the TRUE elements in a Boolean vector:
which(is_old)[1] 1 5 6 8ages_toy[which(is_old)][1] 34 56 41 101This is actually equivalent to ages_toy[is_old], but more explicit, in a sense (sub-setting is done using indices).
3.6.3 Access by name
If you remember well, we saw earlier that it was possible to create vectors where elements are named.
When elements of a vector are named, they can be accessed by their name (represented as a string, i.e. between quotes):
composition <- c(Ca=73, Mg=2, Na=4.5, K=1.3)
composition["Na"] Na
4.5 composition[c("Mg", "Ca")]Mg Ca
2 73 The result is a named vector.
Accessing elements by indices also works:
composition[4] K
1.3 Let’s practice a little more with this useful feature.
Suppose that (for those not interested in PCR mixes), we have a cooking recipe, but that we got the quantities and names of the ingredients separately:
# Recette des cannelés du "professeur Bécavin de la liste bioinfo"
cake_recipe <- c(225, 1, 2, 1, 1, 125, 500)
cake_ingredients <- c("sugar", "butter", "eggs", "yolk", "vanilla", "flour", "milk")It is possible to add names to a vector after creation, assigning a vector of strings to the names, as follows:
names(cake_recipe) <- cake_ingredients
cake_recipe sugar butter eggs yolk vanilla flour milk
225 1 2 1 1 125 500 The names are preserved by some operations. Now, suppose our recipe serves 12 people.
How much flour and milk would we need for 25 people?
3.7 Useful vector-generating functions
3.7.1 Repeating values
The rep function generates a vector by repeating another vector a certain amount of times:
rep(TRUE, times = 3) # same as c(TRUE, TRUE, TRUE)[1] TRUE TRUE TRUErep(1:2, times = 4) # same as c(1:2, 1:2, 1:2, 1:2)[1] 1 2 1 2 1 2 1 2times is an optional argument. By default, the first argument is repeated just once: the default value of this argument is 1. If we want the value of the first argument to be repeated more than once, we need to give a different value to the times argument, with the = sign.
A function can have several optional arguments, all having names enabling us to tell the function unambiguously what argument we want to specify.
(Mandatory arguments often also have names, but the argument order avoids ambiguities.)
This function can be used to repeat each vector element instead of repeating the whole vector, using the each optional argument.
rep(1:2, each = 4)[1] 1 1 1 1 2 2 2 2times and each can even be combined.
How would you generate three times the following “motif”: one pair of 1 followed by one pair of 2 (the equivalent of c(1, 1, 2, 2, 1, 1, 2, 2, 1, 1, 2, 2)) using the rep function?
3.7.2 Sequences of values
We have already seen the n:m syntax to generate numbers from n to m. The seq function offers more control on the way the sequence is to be generated thanks to its by optional argument:
seq(0, 8, by = 2)[1] 0 2 4 6 8The first two arguments are the starting and ending elements. You can also specify them using the to and from argument names. The by argument is the increment. One can use this to generate sequences of non-integral numbers:
seq(-1, 1, by = 0.25)[1] -1.00 -0.75 -0.50 -0.25 0.00 0.25 0.50 0.75 1.00Or, using all arguments’ names:
seq(from = -1, to = 1, by = 0.25)[1] -1.00 -0.75 -0.50 -0.25 0.00 0.25 0.50 0.75 1.003.7.3 Sorting values
If you have a vector containing numbers or strings, it is possible to sort the values using the sort function:
ages_toy[1] 34 4 18 5 56 41 23 101sort(ages_toy)[1] 4 5 18 23 34 41 56 101In the case of a vector of strings, the order is the “lexicographic” order, i.e. using similar sorting rules as in a dictionary:
bikes[1] "Pinarello" "BMC" "Giant" "Cannondale" "Kona"
[6] "Specialized"sort(bikes)[1] "BMC" "Cannondale" "Giant" "Kona" "Pinarello"
[6] "Specialized"There are however some subtleties. For instance, capital letters come before small letters. Strings representing numbers come before letters. This is due to how characters are encoded into numbers for the computer.
sort(c("a", "A", "b", "B", "1", "2"))[1] "1" "2" "a" "A" "b" "B" Be careful when sorting strings containing special characters. The order between special characters may depend on how the computer is set up.
The sort function has a decreasing optional argument. To reverse the order, use decreasing = TRUE
sort(ages_toy, decreasing = TRUE)[1] 101 56 41 34 23 18 5 4sort(bikes, decreasing = TRUE)[1] "Specialized" "Pinarello" "Kona" "Giant" "Cannondale"
[6] "BMC" 3.7.4 Taking decisions using Booleans to make new vectors
Booleans are at the basis of the art of taking decisions.
Suppose we have a grade between 0 and 20 (for instance, 12):
grade <- 12Now we want to decide whether the student who got this grade "passed" or "failed".
The ifelse function allows us to take a decision (i.e. generate a value among several possibilities) based on a Boolean value given as first argument.
The result will be the second argument if the first is TRUE. Otherwise it will be the third.
ifelse(TRUE, 100, 0)[1] 100ifelse(FALSE, 100, 0)[1] 0Use it to generate a character string that will be either "passed" or "failed".
This can also be applied to a vector of grades, and produce a vector of "passed" or "failed" verdicts.
Let’s generate a (uniformly distributed) random sample of 10 grades between 0 and 20, using the sample function:
grades <- sample(0:20, 10, replace = TRUE)
grades [1] 8 13 10 11 17 0 12 20 17 3The first argument is the set of values from which we sample, the second argument is the number of values we want, and replace = TRUE is to allow the same element to be sampled more than once.
And now lets take decisions for all those 10 students at once:
ifelse(grades >= 10, "passed", "failed") [1] "failed" "passed" "passed" "passed" "passed" "failed" "passed" "passed"
[9] "passed" "failed"This operation can be decomposed in two steps:
- Buid a Boolean vector indicating whether grades in the
gradesvector are at least 10 (TRUE) or not (FALSE).
passed <- grades >= 10- Give this Boolean vector as first argument of
ifelse:
ifelse(passed, "passed", "failed") [1] "failed" "passed" "passed" "passed" "passed" "failed" "passed" "passed"
[9] "passed" "failed"4 String manipulations
4.1 Searching strings
The grep function takes two arguments: a pattern and a vector of strings. It returns the indices in the vector where the string matches the pattern:
week <- c("Monday", "Tuesday", "Wednesday", "Thursday", "Friday", "Saturday", "Sunday")
grep("day", week)[1] 1 2 3 4 5 6 7grep("sd", week)[1] 2 3 4This is typically used to extract those strings by vector sub-setting:
week[grep("day", week)][1] "Monday" "Tuesday" "Wednesday" "Thursday" "Friday" "Saturday"
[7] "Sunday" week[grep("sd", week)][1] "Tuesday" "Wednesday" "Thursday" 4.1.1 Grep variants
grep accepts optional arguments.
Use value = TRUE to directly extract the values instead of the indices:
grep("day", week, value = TRUE)[1] "Monday" "Tuesday" "Wednesday" "Thursday" "Friday" "Saturday"
[7] "Sunday" grep("sd", week, value = TRUE)[1] "Tuesday" "Wednesday" "Thursday" Use ignore.case = TRUE to be case-insensitive:
grep("tu", week, ignore.case = TRUE)[1] 2 6There is also another function, grepl, that returns a vector of Booleans (with TRUE where the expression is found, FALSE otherwise) instead of the indices.
Use it to count the number of strings containing "day" in the names of the days of the week.
Now use it to find the days of the week not containing "day".
4.2 Combining strings
We can create longer strings from several strings with the paste function:
paste("I", "can", "paste", "words", "together")[1] "I can paste words together"The above use of paste results in a single string where the given string arguments are separated by spaces (" ").
It is possible to use a different separator to join the strings given to paste, thanks to its sep optional argument:
paste("I", "can", "paste", "words", "together", sep = "")[1] "Icanpastewordstogether""" is an empty string. Here, we tell the function paste to insert nothing between the words (a convenient shortcut to do just that is the function paste0).
This function may look a bit useless for the moment: We could directly type "I can paste words together". However, in more advanced situations, paste can be useful to automatically generate strings (for instance to use in figure legends).
paste can also act on vectors:
paste(c("WT", "WT", "KO", "KO"), c(1, 2, 1, 2), sep = "_")[1] "WT_1" "WT_2" "KO_1" "KO_2"You obtain a vector of strings whose elements result from pairwise concatenation of the corresponding elements from the original vectors.
Use the rep function to create the arguments of paste in the above example.
Value recycling happens when you use vectors of different lengths. This can be useful to quickly generate series of numbered strings, as follows:
paste("sample", 1:3, sep = "_")[1] "sample_1" "sample_2" "sample_3"Here, the first argument is actually a vector with only one element, which is re-used for concatenation with each of the elements from the second argument.
If you want to paste together elements contained in a vector, you can use the collapse argument, as follows:
paste(week, collapse = ", ")[1] "Monday, Tuesday, Wednesday, Thursday, Friday, Saturday, Sunday" You may need to create text where elements are separated by tabulations and having several lines (a very common way to organize scientific data). This can be achieved using special characters for the sep argument: "\t" for the tabulations and "\n" for the new lines.
Suppose we have vectors containing information about gene locations in a genome:
g1 <- c(chrom="I", start="4118", end="10230",
name="WBGene00022277", score=".", strand="-")
g2 <- c(chrom="I", start="10412", end="16842",
name="WBGene00022276", score=".", strand="+")
g3 <- c(chrom="I", start="17482", end="26778",
name="WBGene00022278", score=".", strand="-")(The above represents the genomic location of three genes in the genome of the nematode C. elegans, in BED format.)
Let’s try to make a textual representation of these vectors, as they would appear in a tab-separated file:
# Create some tab-separated lines
header <- paste(names(g1), collapse = "\t")
line1 <- paste(g1, collapse = "\t")
line2 <- paste(g2, collapse = "\t")
line3 <- paste(g3, collapse = "\t")
# Assemble the lines in a multi-line text
# (BED format doesn't use headers)
file_content <- paste(line1, line2, line3, sep = "\n")
file_content[1] "I\t4118\t10230\tWBGene00022277\t.\t-\nI\t10412\t16842\tWBGene00022276\t.\t+\nI\t17482\t26778\tWBGene00022278\t.\t-"4.3 Displaying text
The way the previous piece of text is displayed in the R console is quite literal: the special characters have not been interpreted as such. You can display the text with special characters “interpreted” using cat:
cat(file_content)I 4118 10230 WBGene00022277 . -
I 10412 16842 WBGene00022276 . +
I 17482 26778 WBGene00022278 . -cat not only prints character strings, but can also perform concatenation (as its name suggests), like paste: If you give several strings as arguments, they will be joined using a space (" ") or whatever other string you use as the sep argument:
# Let's add the header this time:
cat(header, line1, line2, line3, sep = "\n")chrom start end name score strand
I 4118 10230 WBGene00022277 . -
I 10412 16842 WBGene00022276 . +
I 17482 26778 WBGene00022278 . - Please do keep in mind that paste creates text, while cat displays text. This is not the same thing at all. If your intention is to have text that you can use within R for other purposes (further combining strings, providing strings as arguments to a function, \(\dots\)), don’t use cat. cat should rather be used to communicate with the world outside your program (such as giving feedback to the user while the program is running, or at the end, to report about the final outcome of the task the program is meant to perform).
5 INTERMISSION: How to get help
5.1 Internal documentation
R contains a documentation system, that you can visualize in the “Help” tab of the bottom-right pane.
To dive in R the documentation:
help.start()Getting help on a particular matter:
?TRUE
?grep
?paste # Note the default value for sep
?sample
?ReservedHelp documentation in R is separated in various sections (“Description”, “Usage”, “Arguments”, “Details”…). Sections such as “Examples” and “See also” are particularly useful when discovering R and extending your R vocabulary.
If you don’t recall exactly the function you are looking for, ?? can make a search for you:
??operators
??syntax
??csv5.2 Describe your environment
You can see all the variable defined in your environment looking at the “Environment” tab of the top-right pane in RStudio, or list them using the ls command:
ls()You can obtain information about your current session (loaded packages with version number, …) using sessionInfo:
sessionInfo()These two commands can be useful to debug cases where unexpected things happen.
When you try to get help from other people, give them the information provided by sessionInfo() This may help them understanding what is happening on your computer.
6 Exporting and importing data
We have briefly seen how to load a data set into R. It is now time to get into more details on this very essential subject.
6.1 A brief overview of tabular formats
Commonly used formats to store tabular data are the following:
- TSV (“tab-separated values”)
- CSV (“comma-separated values”)
- XLSX (Microsoft Excel spreadsheet format since 2007)
If you need to exchange data with bioinformaticians, knowing a few things about these formats may help ensure a smoother communication.
In particular, you need to be aware of fundamental differences between a simple text format and more complex spreadsheet formats.
6.1.1 “Simple text” formats
“Simple text” means that the files using these categories of format are directly encoded using relatively simple and well defined norms meant to represent text (such as ASCII or UTF-8), and can be opened with a general category of tools called text editors.
The basic and essential functionality of a text editor is to change the characters contained in a simple text file. This can be done in more or less sophisticated ways, but that’s all there is to it. Once the file has been saved and closed, it can in principle be opened and edited using any other text editor.
Simple text files have a long history and a wide diversity of applications. For instance, they are used to store the source code of computer programs. As such, an R script should be saved in simple text format, and the editor of RStudio is actually a text editor. It was likely not developed with this goal in mind, but you can easily use it to edit a Python program if you want (the code will not run in the console, of course, but that’s something different).
Text editors are not to be confounded with word processors.
Simple text formats differ by the conventions used to organize the information they contain. Some of them are meant to represent tabular data, in particular TSV and CSV.
6.1.1.1 TSV format
Files in TSV format, as the name suggests, consist in simple text format, where columns are separated by the special tabulation character ("\t"). For instance:
Column1 Column2 Column3
Value1 Value2 Value3(Note the larger intervals between values, due to tabulations, ensuring their alignment with column headers.)
The main strengths of TSV format are the following:
It is simple, and therefore easy to handle in computer programs (among which some widely used and powerful command-line interface utilities).
It is rather unlikely that the
"\t"separator will be needed inside the textual data present in the columns, thereby making it suitable for most simple tabular information.When displayed on a command-line interface, the wide space taken by the
"\t"separator will make the column delimitation more visible than when using commas.
6.1.1.2 CSV format(s)
Files in CSV format are also simple text files, where the column separator is, strictly speaking, the comma (","). For instance:
Column1,Column2,Column3
Value1,Value2,Value3However, this is also sometimes used to refer more generally to other simple text formats, including TSV and CSV2 (where the separator is a semicolon (";")).
In CSV format, textual data is sometimes placed between quotes in order to allow the presence of the column separator character within textual information.
The CSV2 format uses the semicolon instead of the comma as column separator, in order to enable the use of the comma as decimal separator inside columns containing numeric information.
Example using both text between quotes and semicolon separators:
ID;Info;Coeff
"A";"This text field contains spaces, commas and semicolons: ;";0.666.1.2 “Complex” formats
The XLSX format actually consists in a compressed archive containing various files. This enables the storage of complex information, such as formatting properties (colours, fonts), embedding of graphics, file modification history\(\dots{}\) This format was developed for a specific spreadsheet application (Excel), but organized and documented in such a way that other applications could also be made to use it (this was more complicated with the earlier XLS format).
A consequence is that complex spreadsheet applications are needed in order to handle files in this format. Another is that such files tend to take much more storage space than simple text ones.
If you don’t need the advanced features associated with XLSX format, you’ll save disk space and make the work of your bioinformatician colleagues simpler by using TSV or CSV.
6.2 Exporting tables from spreadsheet applications
In spreadsheet applications (such as LibreOffice, OpenOffice, Excel\(\dots\)) you can export your spreadsheets in a format which is bioinformatics friendly such as TSV or CSV.
In your favourite spreadsheet application you should have in the “File” menu a “Save as” or “Export as” option which allows you to export your data to TSV or CSV format.
Simple text formats do not handle multiple spreadsheets at once, so normally a warning will be issued by your application saying that only the current/active spreadsheet will be saved.
You may also voluntarily restrict what you export by selecting a particular zone of the table before actioning the menus. The actual behaviour may depend on the software you are using.
6.3 Checking the exported table
To properly import a table in R, you need to make sure everything is under control. In particular, you should check that you have an answer for each of the following questions:
- What is the column separator?
- Are there column headers?
- Are text fields quoted?
- What decimal separator is used?
- Are there commented lines, and if so, how are comments indicated?
You may have had full control on the export process, in which case the answers should be known to you.
You can also check a posteriori, by opening the exported file. You should be able to do it using TextEdit (on Mac OSX) or Notepad (on Windows), or any text editor of your choice.
If you happen to be skilled enough with command-line utilities, you can of course also use them to get the answers.
6.4 Importing tables in R
6.4.1 Using the graphical interface:
As we saw before, in RStudio we can import both CSV and TSV files using the “Import Dataset” menu and choose “From Text (base)\(\dots\)”. Then check that the options correspond to how your file is formatted.
6.4.2 Using the console:
TSV and CSV files can be imported as data frames using the read.table function. Depending on the actual format, some arguments will have to be changed. There are also shortcut functions more readily suited to a given format, that are very similar to read.table but with different default values for the optional arguments.
You can get information about these functions and their arguments by reading the help page for read.table:
?read.tableSome potentially useful arguments are the following:
sep, to specify the column separator.header, to indicate that the first line contains column headers.quote, to specify the character used to delimit text fields.dec, to specify the character used as decimal point in numbers.skip, to use in case some lines have to be ignored at the beginning of the file.comment.char, to specify one or more characters that indicate comments.row.names, to specify a column number that should be used as row names instead of as a standard data column.stringsAsFactors, to indicate whether text columns should be converted into factors or not.
The read.csv and read.delim variants of read.table assume that the first line contains column headers (header = TRUE) and use a comma (sep = ",") or a tabulation (sep = "\t") as column separators, respectively.
7 Descriptive statistics on vectors
Statistical analysis has several phases, among which:
- Data collection.
- Descriptive statistics (or “exploratory analysis”): characterizing the data.
- Inferential statistics: extrapolating from the data (to estimate confidence intervals, to test hypotheses\(\dots\)).
The third phase will be nourished by information obtained during the second.
In practice, a series of measures (collected for a certain number of samples, for instance) will typically be represented using a vector in R. Those vectors will very often be columns in a data frame reporting several series of measures for the same sampled individuals. For the moment, we will only deal with one vector at a time.
7.1 Samples and random variables
During the collection phase, individuals from a population are sampled. One (or several) characteristic(s) of interest are recorded. The measure of a characteristic of interest varies between individuals. Each one of these observations can be viewed as a realization of a random variable.
For instance, if a mouse is taken from the lab’s collection and its weight is measured, a certain value will be recorded. If another mouse is taken (or the same but at another time), and its weight is measured, a possibly different value will be recorded. This process of selecting a mouse and measuring its weight involves many causes of variability (hence the term “variable”), that are not predictable (hence the qualification as “random”). A sample of values is constituted by the repetition of the same process, and is therefore seen as a series of realization of the same random variable.
Before diving into more theory, let’s do a very practical exercise.
We prepared for you a small table in the form of a XLSX-formatted file. Download it from the link, and try to convert it in a format suitable for loading into R.
Now use the read.csv function to load it as a mice data frame into R. See ?read.csv for some help.
Let’s extract from this table the column containing the sexes of the mice:
mice_sexes <- mice$sexTo get a smaller data frame containing only the data for males, do the following (you will have to wait the chapter about data frames for explanations):
male_tab <- mice[mice_sexes == "M",]We can quickly visualize the sampled weights, sorted using sort:
sort(male_tab$weight) [1] 25.09 25.75 27.09 27.53 27.93 28.35 28.78 28.82 29.67 29.83 32.51We have seemingly random values between \(25\) and \(33\).
7.1.1 Types of random variables
Depending on the nature of the data, the values can fall into different categories of random variables.
The data can be:
- quantitative: Quantities encoded by numbers.
- qualitative: Qualities encoded with arbitrary labels (sometimes called factors in R).
Quantitative random variables can be:
- continuous: Measures falling within a continuous range of numbers (example: size of a person).
- discrete: Countable quantities, corresponding to integral numbers (example: number of children a person has).
Qualitative random variables can be:
- ordinal: A meaningful order can be defined between the different possible labels. This often results from an underlying quantitative variable (example: amount of physical activity labelled using levels such as “low”, “high” or “medium”).
- categorical (also called nominal): There is no meaningful order (example: eye colour).
Note that in a broader sense, qualitative variables are discrete. Indeed, their possible values are not numbers, but they can take a countable number of values.
As a consequence, a number of statistical tools will be equally applicable to discrete quantitative variables and to qualitative variables.
Ordinal qualitative variables are actually quite similar to discrete quantitative variables. It is easy to associate successive integers (1, 2, 3, \(\dots\)) to successive levels of an ordinal qualitative variable.
It is sometimes possible to view the same data in several ways, and have different options to encode them.
male_tab$weight and mice_sexes both are vectors of data. In terms of the categories we just described, what kind of random variable would you associate to each of them?
7.1.2 Population and sample
In statistics, we usually don’t have access to the population we want to study. We only have access to a fragment of it, the sample.

We are going to use the information from this sample to extract (or, more accurately, to infer) general rules about the population. These rules are what is often called a model.
The choice of the model and its parameters will be based on various considerations, including observations made on the samples using descriptive statistics.
7.2 Distributions
All sorts of operations can be done to describe a sample of values, which can easily be executed using functions in R.
7.2.1 Describing quantitative data
7.2.1.1 The histogram
A quick and easy thing to do is to plot a histogram, using the hist function.
We already did it for the ages recorded in the Milieu Intérieur data a long time ago. Let’s do it again:
hist(ages)
The \(x\)-axis displays the range of the values in the studied sample. It is split into non-overlapping contiguous intervals (usually of equal lengths). On the above graph, the intervals are bins of 5 years each, i.e. \([20-25)\), \([25-30)\dots\)
Never plot a histogram for qualitative data! The \(x\)-axis of a histogram is indeed meant to represent a continuous range of numerical values. For discrete data, a barplot is usually more suited (we’ll see how to do this very soon).
The \(y\)-axis represents the amount of data in each one of the intervals, and is labelled “Frequency”.
For finer control on the representation, the breaks optional argument allows us to specify the boundaries of the bins.
Let’s try it using our small mice weights example:
hist(male_tab$weight, breaks = seq(from = 25, to = 33, by = 2))
Here, we made bins of width \(2\), thanks to the by argument of the seq function.
The width of the bins is a compromise between accurately representing the data (thin bins), and effectively summarizing (wide bins).
Here are two other possible choices:
hist(male_tab$weight, breaks = seq(from = 25, to = 33, by = 0.25))
hist(male_tab$weight, breaks = seq(from = 25, to = 33, by = 4))
\(2\) looks like a good compromise.
Let’s look more into details how a histogram is made. The data points are “distributed” in the bins according to their values:
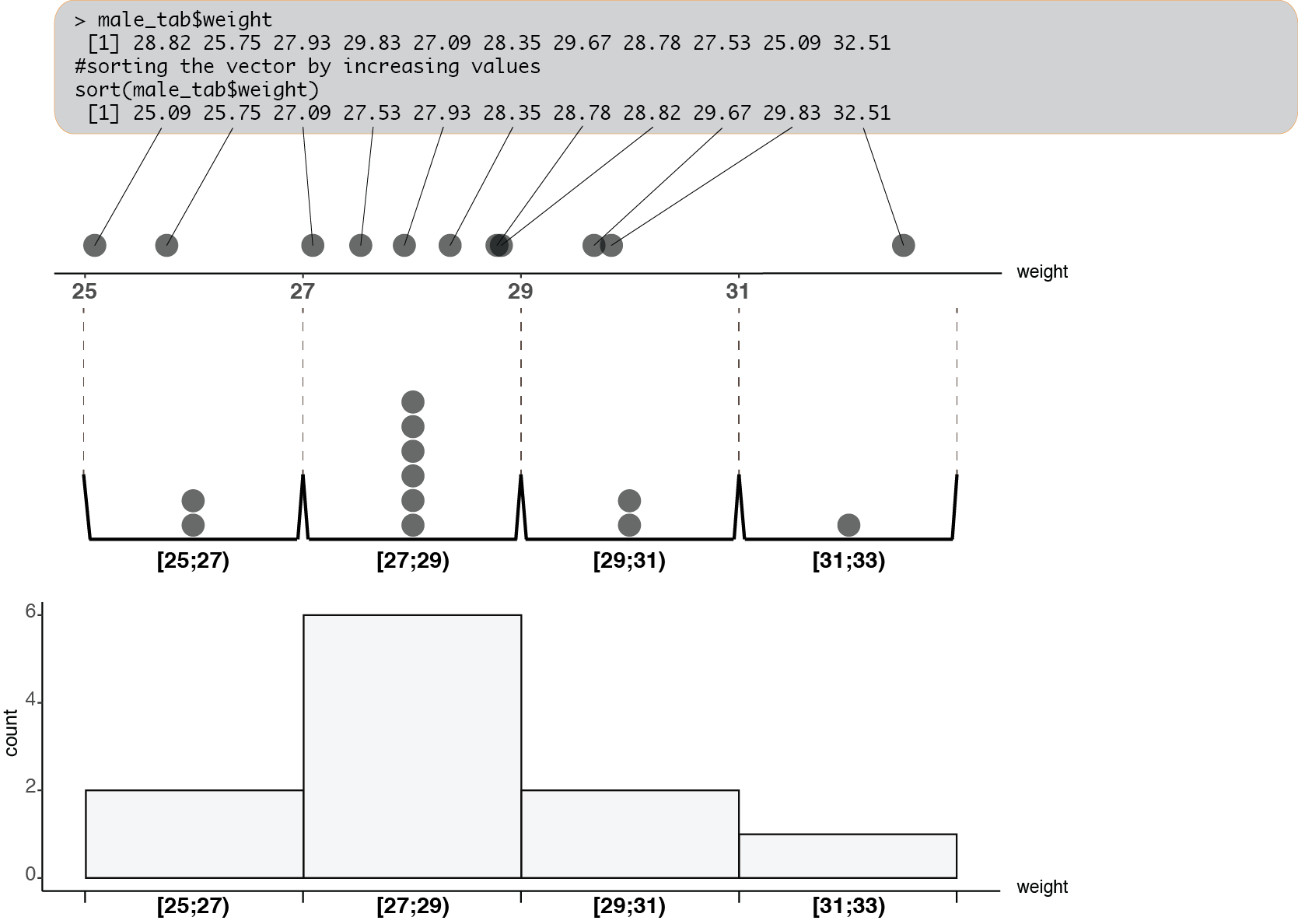
This kind of graphical representation informs us about the distribution of the values in the vector: where values can be found, how broadly they are spread, where they are more concentrated\(\dots\)
Let’s have a quick look at some other measures recorded in our mi data frame.
hist(mi$HeartRate)
hist(mi$Temperature)
The histogram representation reveals striking differences between the distributions of those random variables, and that of the ages.
7.2.1.2 Numerical summaries
A distribution can also be characterized using numbers, such as the mean value, and others. Some of these numbers can be obtained using the summary function, in the form of a named vector:
summary(ages) Min. 1st Qu. Median Mean 3rd Qu. Max.
20.17 35.83 47.71 46.49 58.35 69.75 summary(mi$HeartRate) Min. 1st Qu. Median Mean 3rd Qu. Max.
37.00 54.00 58.00 59.21 65.00 100.00 summary(mi$Temperature) Min. 1st Qu. Median Mean 3rd Qu. Max.
35.70 36.20 36.40 36.43 36.60 37.70 We will soon learn in more details what all these values mean.
We can see that these summary values differ. This reflects differences in the shapes of the histograms and in values on their \(x\)-axes.
The histogram and these summary values are our first step at practicing descriptive statistics.
7.2.2 Describing qualitative data
Now what happens in the case of discrete or qualitative variables?
We can try the summary function on other columns of the data frame:
smoking <- mi$Smoking
summary(smoking)Active Ex Never
161 223 432 sex <- mi$Sex
summary(sex)Female Male
417 399 summary gives different kinds of results, depending on the kind of data it received. Here, the data was encoded as factors, a kind of value in R that we haven’t discussed in details yet. Factors are very well suited to represent qualitative variables. This is what is obtained by default for columns containing text in a table loaded in R.
When given factors summary gives the number of individuals falling into each category, or level.
7.2.2.1 Counting using table
We can obtain the same information (in a slightly different form) using the table function:
table(smoking)smoking
Active Ex Never
161 223 432 table(sex)sex
Female Male
417 399 Contrary to summary, table works the same with any kind of vector, not just factors. It can be used to count Booleans, strings, numbers\(\dots\) Use it!
Values from a table can be extracted using the corresponding labels, like with named vectors:
counts_smoking <- table(smoking)
counts_active <- counts_smoking["Active"]
counts_activeActive
161 counts_inactive <- counts_smoking[c("Ex", "Never")]
counts_inactivesmoking
Ex Never
223 432 How would you obtain the total number of inactive smokers ?
Using table, a contingency table can be created to highlight the interrelation between two variables. In our case, we can create such a contingency table to link the two variables: smoking habits and sex.
table(mi$Sex, mi$Smoking)
Active Ex Never
Female 77 95 245
Male 84 128 187This table displays the number of individuals observed to have a particular combination of factors. This type of table is used to study the association between two kinds of classification (for instance, classification according to the sex, and classification according to the smoking habits). It is used in particular in Fisher’s exact test.
For the contingency table to make sense, the arguments should be vectors corresponding to measures made over the same series of individuals. In particular, the order of the individuals should be the same in all the vectors.
7.2.2.2 The barplot
To visualize the distribution of categorical data, the barplot function can be used with a table of counts. The result is a graph where each category will be represented by a bar:
barplot(counts_smoking)
barplot(table(sex))
Do not use barplots for non-discrete data! Barplots are not histograms:
- The bars are not contiguous.
- The order of the categories on the \(x\)-axis of a barplot can be changed, not the order of the bins in a histogram.

7.2.3 From data distribution to population model
One important use of descriptive statistics is to help us choosing the correct tools for later analyses. One aspect is to characterise our samples in terms of the distribution of its values. The distribution of the values in the sample may then suggest ways to model the distribution of the values in the population. This distribution model will be an essential tool in later phases of statistical analysis.
The word “distribution” can be used for two different but related ideas:
- How the values are distributed in a sample (empirical distribution).
- How the values would be distributed if the random variable from which our sample was observed behaved according to certain rules, to a certain model (theoretical distribution).
In the latter sense, a distribution is characterized in terms of probabilities. What is the probability to observe a given value, or to observe a value within a given range?
We will now present some common ways to obtain a distribution to model a random variable.
7.2.4 Discrete distributions
Discrete distributions will be preferred to model naturally discrete quantities (such as numbers of heads on hydras or numbers of popes at certain times in history) that will be represented using integral numbers. They can also be used for some continuous aspects of the physical world that are typically modelled using discrete classes, such as colours (the spectrum of radio-frequencies is continuous, but it is often more practical to score observations according to a discrete set of colours).
A model for a discrete distribution would consist in associating each possible value with the probability of obtaining it when randomly sampling an element from the population.
For instance, the “Milieu Intérieur” data frame contains information regarding individual’s relationships with smoking. This is scored using three categories (a factor with three levels, in R terminology):
Active, for active smokers;Ex, for people that used to smoke but no longer do;Never, for people that never got into smoking.
If we wanted a model for this random variable, we could associate each category with a number, between \(0\) and \(1\), and such that the three numbers sum to \(1\) (this is a required property for probabilities). Determining the values for these numbers could be done in various ways (using a priori knowledge, estimating the values from the sample, \(\dots\)).
For instance, we could just use the proportions of each category in the sample as probabilities for our model. This can be computed as follows:
counts_smoking <- table(smoking)
counts_smokingsmoking
Active Ex Never
161 223 432 nb_observations <- sum(counts_smoking)
nb_observations[1] 816proportions_smoking <- counts_smoking / nb_observations
proportions_smokingsmoking
Active Ex Never
0.1973039 0.2732843 0.5294118 Besides using such empirical probabilities to build a model, there are various well studied classical discrete distributions available: Bernoulli, binomial, uniform, Poisson\(\dots\)
Their ability to be mathematically described reflect some “rules”, some regularities or mechanisms, in the process underlying the random variable to which they are associated. This is what makes them useful to extrapolate beyond simple empirical probabilities. Indeed, empirical probabilities will fit the sampled data perfectly, but these probabilities will likely not reflect what happens at the whole population level (especially if the sample is small), resulting in poor behaviour in later phases of statistical analyses. This is sometimes called “over-fitting”.
We will now briefly present a few classical and simple discrete probability distributions.
7.2.4.1 Uniform distribution
This represents the probabilities of equally probable discrete events. Under such a distribution, if there are \(N\) mutually-exclusive events, each one has a probability of \(\frac{1}{N}\). The only parameter needed to use this distribution as a model is \(N\), the number of possible events.
For instance, the outcome of the throw of a standard dice is quite well modelled by a uniform distribution with 6 equally probable events (that we can label using the numbers \(1\) to \(6\)):

7.2.4.2 Bernoulli distribution
This represents the probabilities of a pair of mutually exclusive events. This very simple model has one parameter: the probability of one of the events (that is typically noted \(p\)). The probability of the other event is then \(1 - p\).
For instance, if we are interested in the probability of left-handedness, someone randomly drawn from a population could be either left-handed (\(L\)) or not (\(\bar{L}\)).
The following would be the distribution of probabilities if the probability of left-handedness was \(\frac{1}{10}\):

If we wanted a model for the Sex column of our mi data frame using a Bernoulli distribution, we could use the proportion of females as parameter p. Compute this proportion using the sex vector.
7.2.4.3 Binomial distribution
This represents the probabilities of achieving a certain number of times a given outcome in a series of drawings from the same Bernoulli distribution. This model takes two parameters: the number of drawings, and the probability of the outcome of interest in the underlying Bernoulli distribution.
For instance, assuming a coin flip can either result in a head or a tail, with equal probability (and nothing else), the probability of obtaining a head would be \(\frac{1}{2}\). (This is a case of discrete uniform and Bernoulli distribution at the same time.)
The probabilities of obtaining a certain number of tails in 5 coin flips can therefore be modeled using a binomial distribution with 5 events:

7.2.5 Continuous distributions
Physical measures such as sizes, weights, or durations are continuous by nature. Assuming an arbitrarily high precision, they could take any value between a pair of (possibly infinite) real numbers.
For such measures, each value is one possibility among an infinity of others. It is therefore not useful to associate individual probabilities to each possible value because the probability of one value among an infinity of other possibilities is infinitesimal.
A model for a continuous random variable should instead associate probability densities to real numbers in the (possibly unbound) range of possible values for that variable, in the form of a probability density function.
Let’s take again our male mice weights example and try to figure out what a probability density is.
As we have seen with the smoking habits example, a probability can be estimated from empirical data by taking the proportions of observations within each category. The same can be done with continuous data, using histogram bins instead of categories.
Now, a probability “density” can be derived from these per-bin probability estimates, by scaling them by the bin widths.
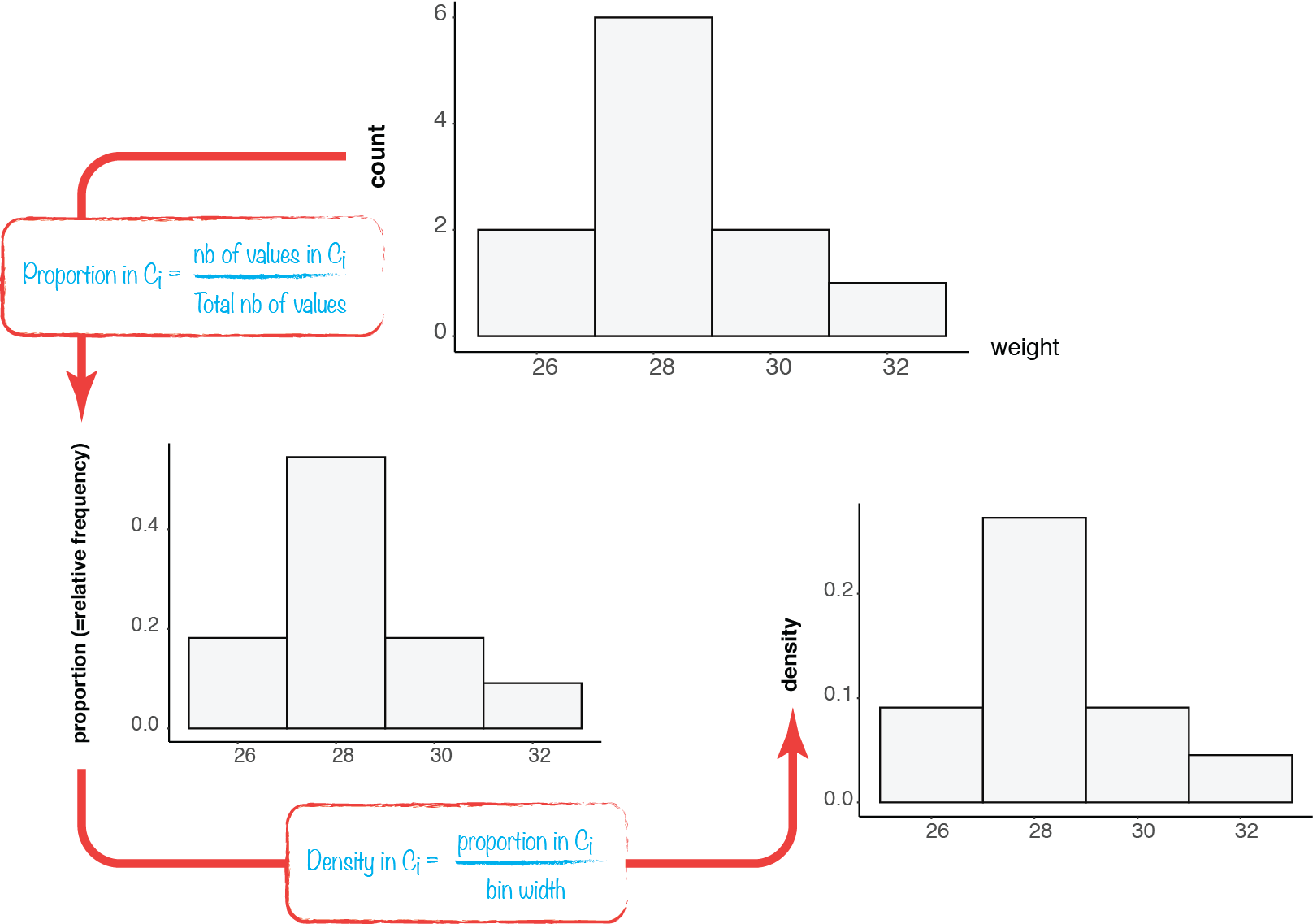
Here, we transform the probabilities of certain ranges of weights into a kind of probability “per unit of weight”.
Numerical analyses techniques exist that can smooth these densities to extrapolate a distribution for the underlying population. This is a way to provide an empirical model of continuous distribution for the population.
In R, the density function can make such an estimation of densities, which can be plotted using the plot function:
plot(density(male_tab$weight))
(The plot function is a versatile function that tries to guess what kind of graph we want based on the nature and numbers of arguments.)
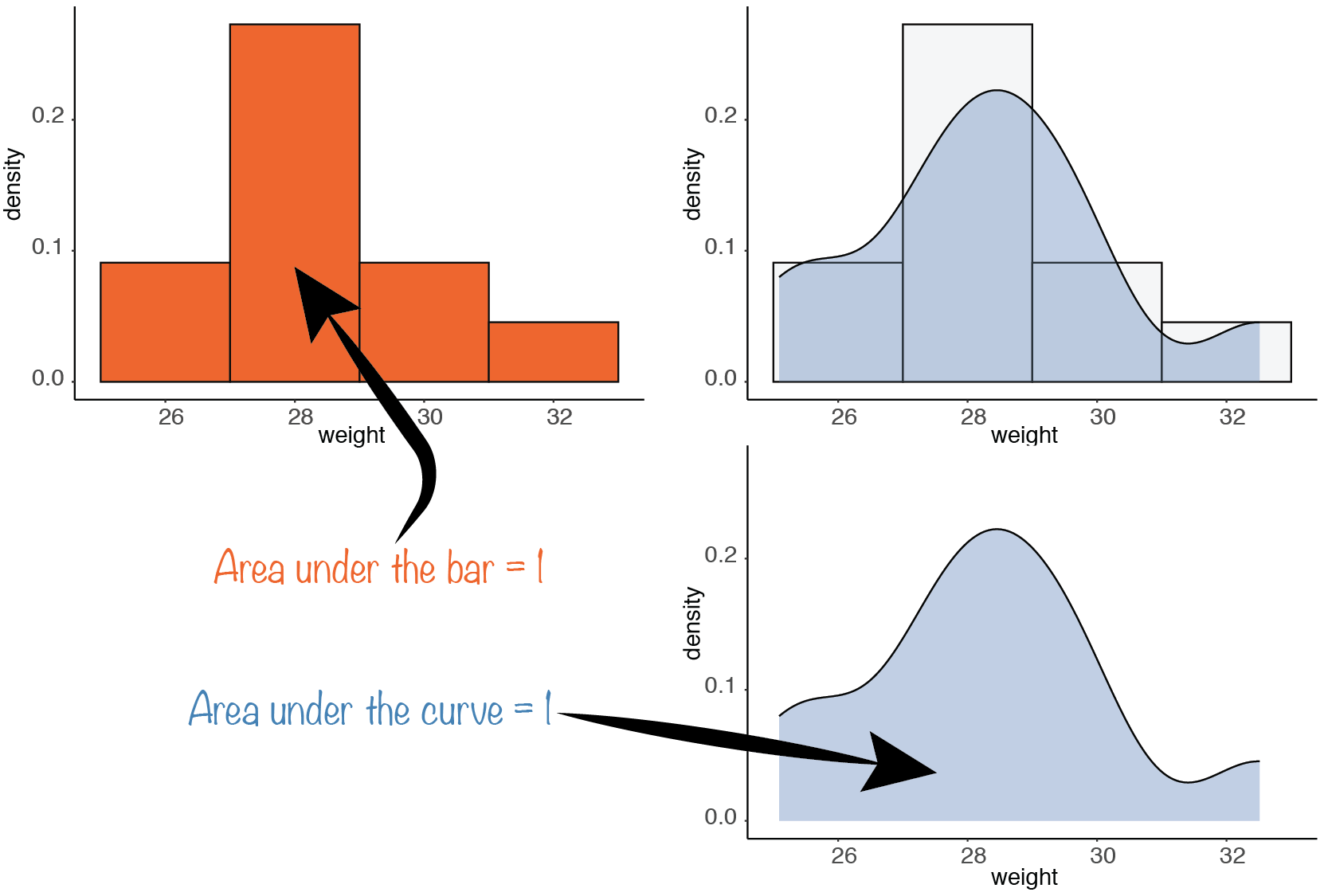
An important property is verified for both the smooth and non-smooth densities: The total area under the curve (sum of the areas of the bars, in the non-smooth case) is \(1\).
In mathematical terms, a probability density function is a positively-valued function that, once integrated between two values of its domain of definition, will give the probability to sample a value between these two boundaries. Therefore, the area below the whole curve corresponds to the probability that a sampled value will be “somewhere where values are possible”, which is \(1\).
Mathematical integration of a function can be visualized on the function’s graph as the area under the curve between the boundaries:

As we can see, on the above example, for a same interval width (\(2\)), the probability of a value between \(-4\) and \(-2\) is much lower than the probability of having a value between \(0\) and \(2\). This reflects the fact that the above probability density is higher around zero than far from it.

As was the case for discrete distributions, some classical continuous distributions exist. For instance: Continuous uniform, Gaussian (or Normal), Chi-squared, Student, Fisher\(\dots\)
They may prove helpful when trying to generalize beyond experimentally derived probability densities.
We will now briefly present a few of them.
7.2.5.1 Continuous uniform distribution
This distribution is characterized by an equal probability density all along a given interval. It is characterized by a \(min\) and a \(max\) parameters, delimiting that interval.
Here is the curve of the density function for the continuous uniform distribution on the \([-1; 1]\) interval:

The density is easy to express in terms of the \(min\) and the \(max\) parameter: \(\frac{1}{max - min}\).
Check that this is the case on the above figure, and compute the area of the rectangle delimited by the density curve and the \(x\)-axis.
7.2.5.2 Normal distribution
This is arguably the most famous family of continuous probability density functions. It is also known as “Gaussian” (from Gauss, a scientist who discovered it trying to characterize the distribution of measurement errors). It is sometimes also qualified as “Bell shaped”, for the shape of the curve of its probability density function:

This distribution is symmetrical takes two parameters, the mean (determining around what value it is centered) and the standard deviation (determining how spread it is around the mean). We will discuss those terms with more details later.
The above curve is the one for the standard normal distribution, that is, the one whose mean is \(0\) and standard deviation is \(1\).
Here is what the curves look like with mean \(0\) and standard deviations \(0.5\), \(1\) and \(2\):

If a random variable is modelled using a normal distribution, it is often useful to transform the data so that the standard normal distribution can be used.
This standardization can be done by simply subtracting the mean of the values, so that the values are centered around zero, and then dividing by the standard deviation of the values (more about this soon), so that the standard deviation now becomes \(1\). This can be called a scaled variable. (For French speakers, this is called “centrer et réduire”, and the result is a “variable centrée réduite”.)
This kind of transformation can be applied even if the variable does not follow a normal distribution. Just keep in mind that the result will not follow a standard normal distribution, in that case.
7.2.5.3 Student’s t-distribution
This distribution was discovered by William Sealy Gosset, an employee of the Guinness brewery who published his work under the pseudonym “Student”.
Like the standard normal distribution, it is bell shaped and centered around zero. However, it is more widely spread.
It takes a parameter called “degree of freedom”, which is an integral number. The higher the degree of freedom, the lower the dispersion of the values, and the closest to the standard normal distribution.
Here is a Gaussian distribution, together with t-distributions of various degrees of freedom:

7.2.5.4 Chi-squared distribution
The Chi-squared distribution (or \(\chi^2\)-distribution) also has a parameter named “degree of freedom”. If \(df\) is the degree of freedom, the particular distribution can be noted \(\chi_{df}^2\), and is the distribution of the sum of \(df\) squared independent random variables following the standard normal distribution.
For instance, if a variable is modelled using a normal distribution, centering the values (by subtracting the mean), then normalizing (by dividing by the standard deviation), and finally squaring, would result in a random variable that is expected to follow a \(\chi_{1}^2\) distribution. This has various applications.
As a sum of squares, this probability density is only defined for positive numbers. Here are the curves for various degrees of freedom:

7.2.6 Vectors of random values
It can sometimes be useful to generate data following certain probability distributions.
R provides a family of functions whose name starts with “r”, and then something reminding the name of a theoretical distribution: rbinom for the binomial distribution, rnorm for the normal distribution, rt for Student’s t-distribution, rchisq for the Chi-squared distribution\(\dots\)
These functions generate vectors of random values drawn from the corresponding distribution.
For instance, the rnorm function generates a vector of random values from a standard normal distribution (mean 0 and standard deviation 1):
ten_norm <- rnorm(10)
ten_norm [1] -0.4764353 1.5712980 -0.5405243 -0.5895441 1.0954162 -0.6731219
[7] 0.1096119 1.0659272 0.1661113 0.2101096The argument is the number of values to draw from the distribution.
We can quickly visualize the distribution of the obtained numbers using the hist function:
hist(ten_norm)
We can change the parameters of the normal distribution using the mean and sd optional arguments. mean and sd have default values of 0 and 1 respectively, so that rnorm draws from the standard normal distribution by default.
For instance, we can generate values from a normal distribution of mean \(10\) and standard deviation \(0.1\) as follows:
ten_norm_2 <- rnorm(10, mean = -4, sd = 0.1)
ten_norm_2 [1] -3.934045 -4.089639 -4.163901 -3.755335 -3.892571 -3.983323 -3.940167
[8] -4.180485 -4.114692 -3.950806The histogram should reflect the change in parameters:
hist(ten_norm_2)
Use the help of the rbinom function (?rbinom) to find how to generate 32 random values drawn from a binomial distribution modeling the number of tails obtain when flipping five coins. Create a flip_5_32 vector and make a barplot to represent the obtained distribution.

7.3 Location
One of the aspects by which a distribution of values can be characterized is its “central tendency”, or “location”. This is a way to estimate what a “typical” value from that distribution would be. There are several approaches to do this kind of estimation. The most commonly used location parameters are the mean and the median.
7.3.1 Arithmetic mean
The best known location descriptor is the (arithmetic) mean (or “average”) of the values. For a sample of values, it can be computed as the sum of the values, divided by the number of values.
The mean \(m\) of a sample with \(n\) values (noted \(x_1\), \(\dots\), \(x_n\)) can be expressed as follows:
\[m = \frac{x_1 + ... + x_n}{n}\] or (using the mathematical notation for a sum): \[m = \frac{\sum_{i=1}^{n}x_i}{n}\]
When using a theoretical distribution to model a population, the theoretical distribution has to be tuned using parameters. These parameters are often noted using Greek letters.
For instance, to have the theoretical distribution centered around the same values as the data, one would typically use the mean as a tuning parameter. The (unknown) mean of the population would be the ideal value for the mean parameter (noted using the Greek letter \(\mu\)). It will be estimated using the (known) sample mean (\(m\)). Such an estimated mean is sometimes noted \(\hat{\mu}\), to distinguish it from the real population mean \(\mu\). \(\hat{\mu} = m\). If the sample is not biased, \(\hat{\mu}\) for large samples will tend to be close to \(\mu\).
Estimators obtained from a sample are often noted with decorations such as \(\hat{\phantom{\mu}}\) to distinguish them from the parameters of mathematical models chosen to represent probability distributions of the population.
Can you compute the mean of the values in the ages vector?
We do not need to manually compute the mean using sum and length. There is actually a mean function for that:
mean(ages)[1] 46.48571We can try to apply mean on a vector of Booleans:
mean(ages > 30)[1] 0.8504902 What does this value represent?
Can you find the mean of the ages, excluding young people (assuming old age starts at 64)?
7.3.2 Median
Another descriptor for the location of a distribution is the median, that is, the value that separates the distribution in two halves of equal numbers of elements.
Two cases may be distinguished:
- If there are an odd number of values, the median is the middle value.
- If there are an even number of values, one usually takes the average of the two middle values.
Determine the median of our small ages_toy vector.
Reminder:
ages_toy <- c(34, 4, 18, 5, 56, 41, 23, 101)Contrary to the arithmetic mean, the median cannot be computed using a simple mathematical formula. It is a bit painstaking to compute a median “manually”. Fortunately, R provides the median function, that does all the tedious work of searching this value for us:
median(ages)[1] 47.71An important property of the median is that it is less sensitive than the mean to the presence of atypical values (“outliers”).
To illustrate this, let’s consider again our small vector of ages:
ages_toy[1] 34 4 18 5 56 41 23 101Suppose that our data was obtained from a table where a reporting error occurred, so that we instead obtained this vector:
ages_wrong <- c(34, 4, 18, 5, 560, 41, 23, 101)If a value above the median is erroneously increased, or a value below the median is erroneously decreased, the median will not be affected.
The mean, however, will change:
median(ages_toy)[1] 28.5median(ages_wrong)[1] 28.5mean(ages_toy)[1] 35.25mean(ages_wrong)[1] 98.25Here is another small example, this time with an odd number of elements. The median is indeed one of the elements:
powers <- c(1, 2, 4, 16, 256)
median(powers)[1] 4Notice how “skewed” the above distribution is. The median is much closer to the minimum than to the maximum value. What about the mean?
mean(powers)[1] 55.8The above example shows that a location estimator is far from enough to characterize a distribution, and may sometimes not be very useful.
7.3.3 Mode
The mode of a distribution of value is the most frequent value, the most likely to be sampled.
Considering the mode as a location parameter most makes sense for densely sampled discrete distribution. For small samples of values taken from a large or a continuous range, no meaningful mode is likely to emerge, as most sampled values will be unique. However, if the global population from which such a sample is obtained is modeled using a continuous distribution, the theoretical mode can be meaningful and will be the value at which the corresponding probability density is highest.
There is no R function to compute the mode (at least by default). We will soon see how to compute the mode of a sample of values.
7.4 Dispersion
The location of a distribution of values is important to estimate, but distributions with the same location can actually be very different. For instance, the values can be all close to the location parameter, or on the contrary vastly dispersed. This will have an important influence on the outcome of statistical tests.
7.4.1 Range
One very simple way to describe the dispersion of the data is to extract its extreme values:
min(ages)[1] 20.17max(ages)[1] 69.75range(ages) # min and max in one function[1] 20.17 69.75 The range function in R gives the extreme values from a distribution, whereas in statistics, the term “range” is used to refer to the breadth of the distribution, that is, the difference between the highest and the lowest value:
range_ages <- max(ages) - min(ages)
range_ages[1] 49.58It is also possible to use the diff shortcut function:
diff(range(ages))[1] 49.58 Can you compute the index (or indices) of the lowest value (or values) in the age vector?
min, max and range are obviously very sensitive to the presence of outliers.
Find the mode of the smoking vector.
7.4.2 Variance and standard deviation
The variance is a measure of dispersion with respect to the mean. It represents a “typical” value for the (squared) distances to the mean.
The variance \(s^2\) of a sample (of size \(n\)) can be computed as the arithmetic mean of the squared deviations of its values (noted \(x_1\), \(\dots\), \(x_n\)) from its mean \(m\).
\[s^2 = \frac{(x_1 - m)^2 + \dots{} + (x_n - m)^2}{n}\] or: \[s^2 = \frac{\sum_{i=1}^{n}(x_i - m)^2}{n}\]
Some theoretical distributions will have a variance (\(\sigma^2\)) tuning parameter, to make the values more or less spread around the mean.
If we had access to the full population, its variance would be the perfect value for this parameter. However, we only have access to the variance of a sample.
Unfortunately, unless the sample mean happens to be exactly the real (and unknown) population mean, the sample variance will be an under-estimation of the population variance. This comes from the fact that the mean of a sample is the value that minimizes the sum of squared distances to the sampled values.
To estimate the population variance, a slightly modified formula should be used:
\[\hat{\sigma^2} = \frac{(x_1 - m)^2 + \dots{} + (x_n - m)^2}{n - 1}\] or: \[\hat{\sigma^2} = \frac{\sum_{i=1}^{n}(x_i - m)^2}{n - 1}\]
This is the formula implemented in the var function in R:
sum((ages - mean(ages)) ^ 2) / (length(ages) - 1)[1] 191.9444var(ages)[1] 191.9444The two formulas will give close values for large sample sizes, reflecting the fact that population mean estimations will tend to be more accurate using large samples.
You should use the second one when you need a variance parameter to tune a theoretical distribution. That is why this is the formula implemented in var.
The standard deviation can be computed by taking the square root (sqrt function in R) of the variance. This provides a dispersion estimation in the same unit as the studied values, and can be directly obtained using the sd function.
sqrt(var(ages))[1] 13.8544sd(ages)[1] 13.8544Greek letter \(\sigma\) (“sigma”) stands for “standard deviation” in the notation for the variance (\(\sigma^2\)).
Now that you know how to compute a mean and a standard deviation, create a vector representing a standardization of the weights of the male mice.
7.5 Quantiles
For a given distribution, any probability can be associated to a quantile. The quantile at probability \(p\) (or quantile of order \(p\)) is the value \(Q\) such that a proportion \(p\) of the values in the distribution of interest are below or equal to \(Q\). Hence, there is a proportion \(1 - p\) of the values in the distribution that are equal or above of \(Q\).
We have already seen an example of quantile: The median of a distribution is its quantile at probability \(0.5\).
Indeed, the median divides a distribution in two halves with equal number of points. The proportion of the values that are below the median is \(0.5\), and the proportion of the values that are above is \(1 - 0.5\).
Some other widely used quantiles are the first quartile (at probability \(0.25\)) and the third quartile (at probability \(0.75\)). Together with the median (which is the second quartile), they cut the distribution into four bins of equal number of counts.
If we have a density for our population (for instance estimated using the density function), we can have an estimation for the value of any quantile. This can be graphically represented on the plot of the density, as follows:
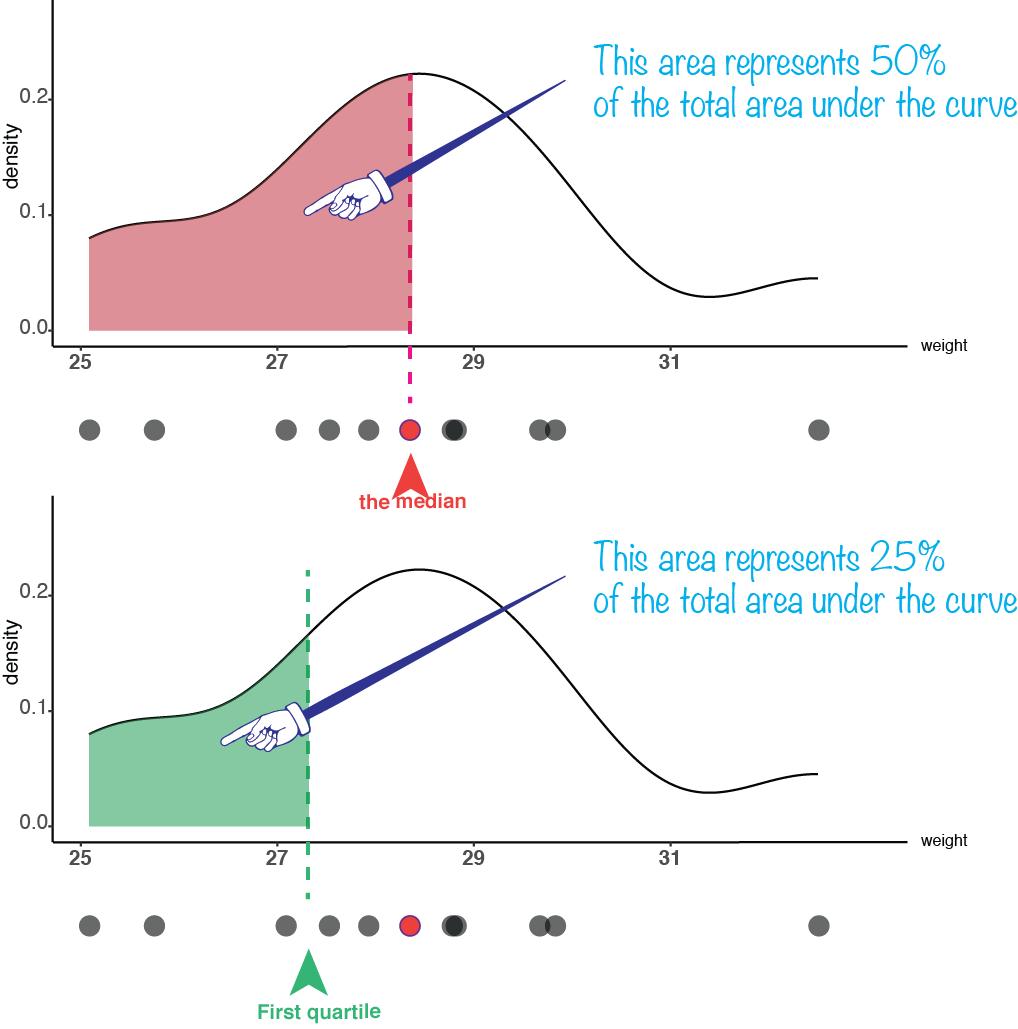
The quantile at a given probability can be estimated using the quantile function:
quantile(male_tab$weight, probs = 0.25) 25%
27.31 quantile(male_tab$weight, probs = 0.5) 50%
28.35 The probs argument specifies the probability whose quantile is desired. It can actually take a vector of probabilities:
quantile(male_tab$weight, probs = c(0.25, 0.5)) 25% 50%
27.31 28.35 The result is a vector containing the cut values, with names displayed above those values. The names correspond to the proportion of the values that are below the corresponding quantile.
When a quantile does not correspond to an actual value in the data, several methods exist to choose a value between the two closest data points (taking the average is the simplest one). This may result in differences between tools. For instance, the boxplot function, that we will present soon, uses a different method than the quantile function.
The distribution of values in a sample can be summarized by determining what values cut the distribution into bins of equal size (in terms of numbers of sampled values falling in the bins). These cutting values are quantiles of the distribution corresponding to regularly spaced probabilities.
The cutting of a distribution into quantiles can be more or less fine-grained, depending on the number of bins into which the distribution of values is cut: quartiles, deciles, centiles.
7.5.1 Quartiles
A typical usage is to cut the distribution into four bins and the corresponding quantiles are then called quartiles.
This is what the quantile function reports when the probs argument is not set:
quantile(ages) 0% 25% 50% 75% 100%
20.1700 35.8300 47.7100 58.3525 69.7500 In the above example:
- 0% of the values are below 20.17
- 25% of the values are below 35.83
- 50% of the values are below 47.71
- 75% of the values are below 58.3525
- 100% of the values are below 69.75
This named vector can be handled just like a normal vector (the extracted values will keep the associated names, though):
age_quartiles <- quantile(ages)
age_quartiles["50%"] 50%
47.71 As mentioned before, the 50% quantile actually corresponds to the median (it divides the distribution in two halves).
The 0% and 100% quantiles should correspond to the min and max.
Extract a subset of age_quartiles containing only these two extreme values.
As mentioned earlier, the behaviour of the summary function depends on the type of data on which it is applied. In the case of a vector of numbers, it computes the quartiles (with fancier names than when quantile is used) as well as the mean:
summary(ages) Min. 1st Qu. Median Mean 3rd Qu. Max.
20.17 35.83 47.71 46.49 58.35 69.75 7.5.2 Interquartile range (and outlier detection)
Since the min and the max are highly sensitive to the presence of outliers, the only quartiles actually considered in practice are those delimiting 25%, 50% and 75% of the distribution. They are sometimes noted \(\textrm{Q}_1\), \(\textrm{Q}_2\) and \(\textrm{Q}_3\), and the difference between \(\textrm{Q}_3\) and \(\textrm{Q}_1\) is used as a dispersion measure named interquartile range (\(\textrm{IQR}\)): \(\textrm{IQR} = \textrm{Q}_3 - \textrm{Q}_1\)
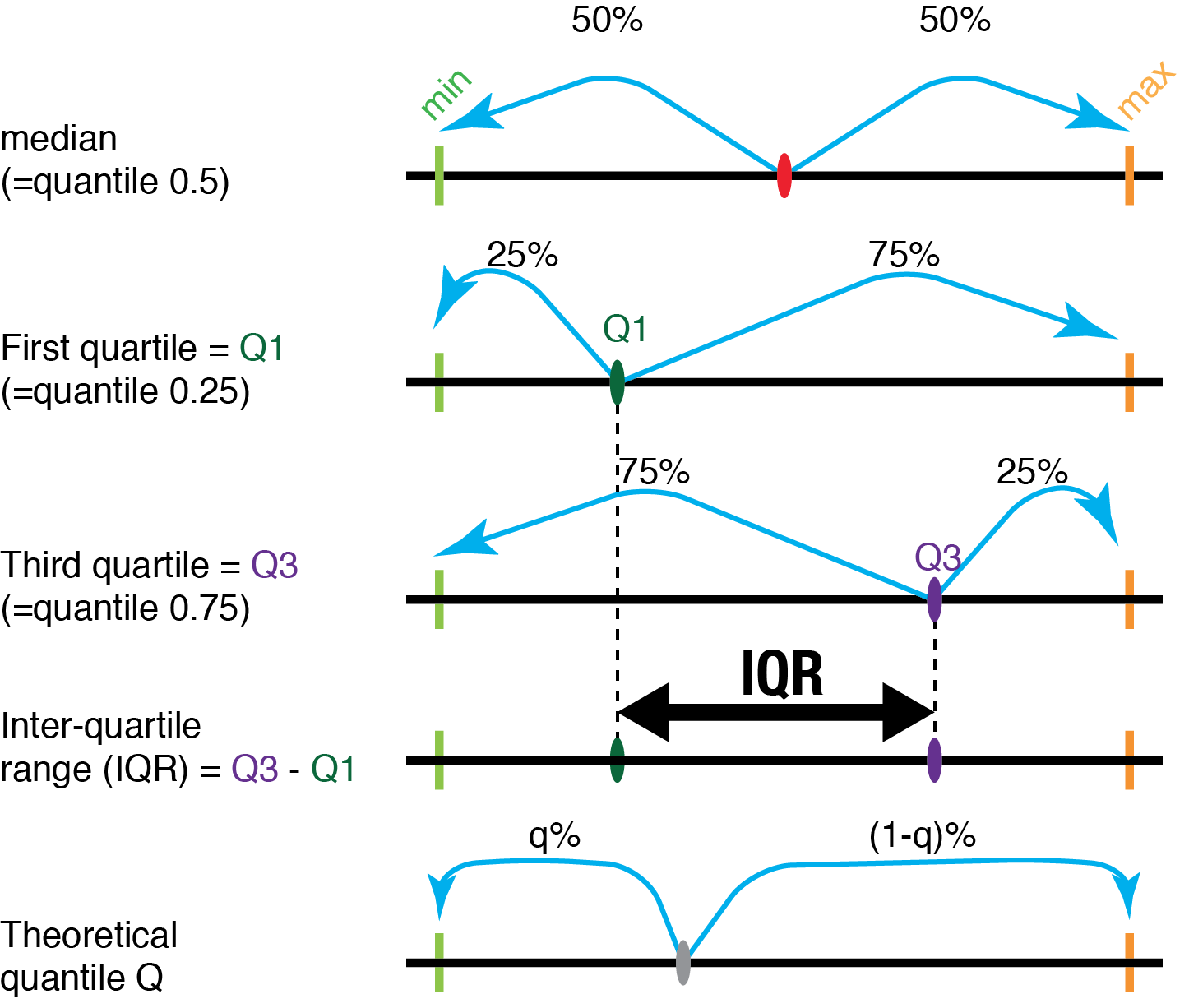
Compute the interquartile range of our ages vector (and put this in a variable named iqr_ages).
Guess what? There’s actually an R function to easily get the interquartile range of a vector:
IQR(ages)[1] 22.5225Like the standard deviation, the interquartile range is in the same unit as the values. It is less sensitive to the presence of outliers, and is actually used to define a criterion to detect outliers. Typically, values below \(\textrm{Q}_1 - 1.5 \times \textrm{IQR}\) or above \(\textrm{Q}_3 + 1.5 \times \textrm{IQR}\) are considered outliers.
Using this criterion, filter out outliers from our ages_wrong small vector example, into an ages_better vector.
Reminder:
ages_wrong <- c(34, 4, 18, 5, 560, 41, 23, 101)Note that the interquartile range will vary depending on the method used to determine quantiles. Outlier detection will therefore also vary.
7.5.3 Boxplots
The median, quartiles and outliers are represented on graphs called boxplots.
Here is a nice boxplot for our mice weights data, with detailed annotations:
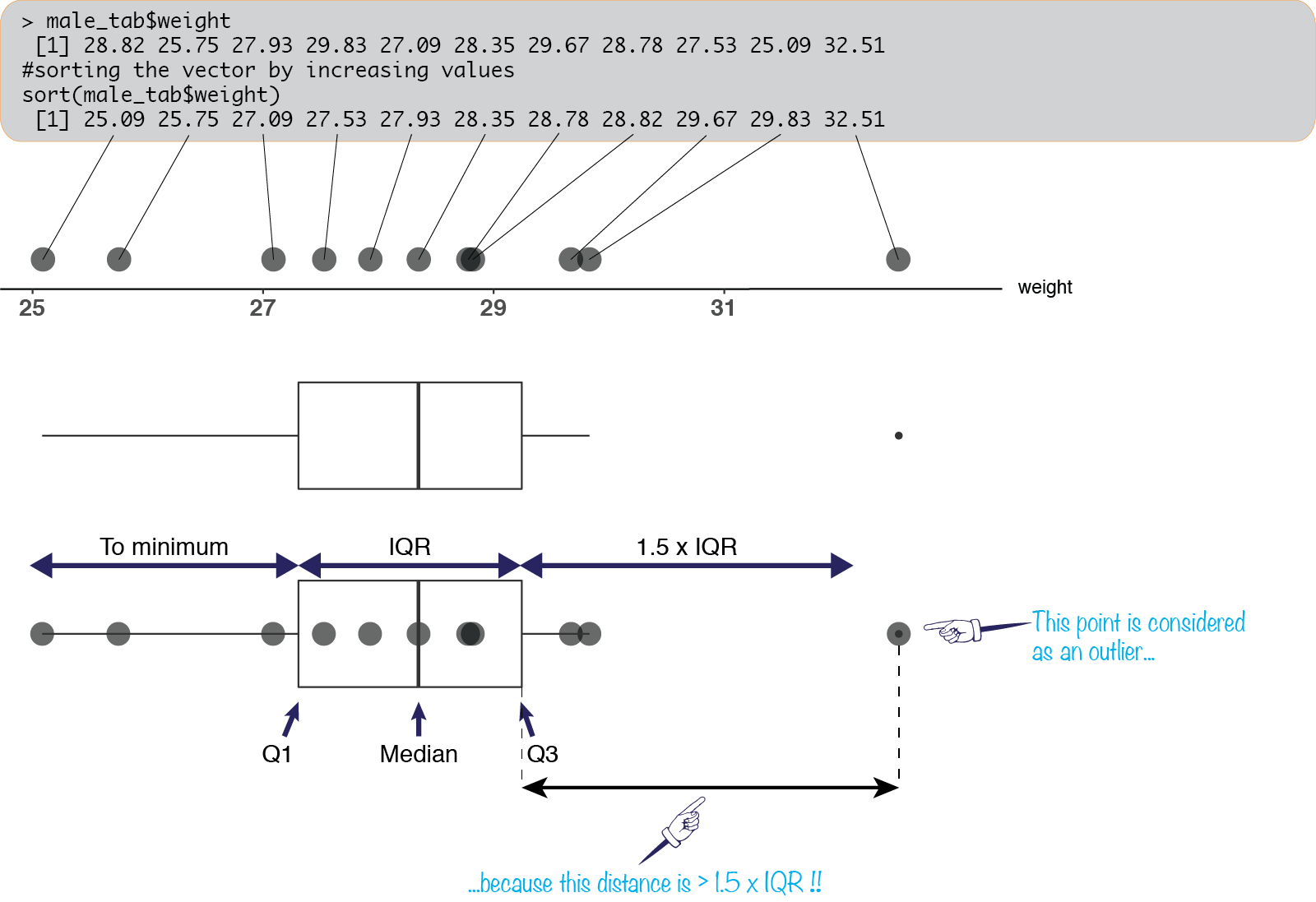
We can make our own boxplots using the boxplot function.
Let’s use it, together with another function, abline, that can add straight lines to a graph:
quartiles_toy <- quantile(ages_toy)
Q0_toy <- quartiles_toy["0%"]
Q1_toy <- quartiles_toy["25%"]
Q2_toy <- quartiles_toy["50%"]
Q3_toy <- quartiles_toy["75%"]
Q4_toy <- quartiles_toy["100%"]
iqr_toy <- IQR(ages_toy)
lower_ok_toy <- Q1_toy - 1.5 * iqr_toy
upper_ok_toy <- Q3_toy + 1.5 * iqr_toy
boxplot(ages_toy,
ylim = c(
min(lower_ok_toy, Q0_toy),
max(upper_ok_toy, Q4_toy)))
abline(
h = Q0_toy,
col = "grey", lty = "dashed")
abline(
h = lower_ok_toy,
col = "grey")
abline(
h = Q1_toy,
col = "green")
abline(
h = Q2_toy,
col = "red", lty = "dashed")
abline(
h = Q3_toy,
col = "blue")
abline(
h = upper_ok_toy,
col = "grey")
abline(
h = Q4_toy,
col = "grey", lty = "dashed")
The h argument of abline is used to indicate the \(y\)-axis coordinate of a horizontal line to draw. There is also a v argument, to specify the \(x\)-axis coordinate of a vertical line. Many plotting functions can also take optional arguments to set “graphical parameters”, such as col for the colour, or lty for the line type. See ?par for more information about graphical parameters.
We used abline to place quartiles and outlier detection limits.
Here, we also use the ylim argument of boxplot, which sets the limits for the \(y\)-axis. We did this to ensure that all these horizontal lines appear in the figure.
There are a few things to comment on the above graph.
- The middle line of the box corresponds to the median (\(\textrm{Q}_2\), dashed red line).
- The upper and lower borders of the box, sometimes called “hinges”, correspond to the first and third quartiles (\(\textrm{Q}_1\) and \(\textrm{Q}_3\)), but using a different calculation method than the
quantilefunction (as can be seen by the non-matching green and red lines). - The “whiskers” correspond either to the “reasonable” extreme values, that is, the lowest or highest values that do not fall beyond the outlier detection thresholds (determined using the \(1.5 \times \textrm{IQR}\) method)
For this data, there are no outliers detected by the boxplot function, but the top grey line, computed based on the quantile function, indicates that with a different way to compute quantiles, the \(101\) years old person (at the top whisker) could be considered an outlier.
Now let’s try with data for which outliers should more surely be detected:
quartiles_wrong <- quantile(ages_wrong)
Q0_wrong <- quartiles_wrong["0%"]
Q1_wrong <- quartiles_wrong["25%"]
Q2_wrong <- quartiles_wrong["50%"]
Q3_wrong <- quartiles_wrong["75%"]
Q4_wrong <- quartiles_wrong["100%"]
iqr_wrong <- IQR(ages_wrong)
lower_ok_wrong <- Q1_wrong - 1.5 * iqr_wrong
upper_ok_wrong <- Q3_wrong + 1.5 * iqr_wrong
boxplot(ages_wrong,
ylim = c(
min(lower_ok_wrong, Q0_wrong),
max(upper_ok_wrong, Q4_wrong)))
abline(
h = Q0_wrong,
col = "grey", lty = "dashed")
abline(
h = lower_ok_wrong,
col = "grey")
abline(
h = Q1_wrong,
col = "green")
abline(
h = Q2_wrong,
col = "red", lty = "dashed")
abline(
h = Q3_wrong,
col = "blue")
abline(
h = upper_ok_wrong,
col = "grey")
abline(
h = Q4_wrong,
col = "grey", lty = "dashed")
7.5.4 Deciles and centiles
A finer-grained description of a sample of values can be obtained by using ten or even a hundred bins when computing quantiles, resulting in deciles and centiles (or “percentiles”) respectively.
This can be computed in R using the probs argument of the quantile function. To obtain deciles, this argument should be set using cumulated proportions from \(0\) to \(1\) by increments of \(\frac{1}{10}\) instead of the default \(\frac{0}{4}\), \(\frac{1}{4}\), \(\frac{2}{4}\), \(\frac{3}{4}\) and \(\frac{4}{4}\).
Generate a decile_cumul_props vector containing these proportions.
We can now use this vector to obtain the deciles of our ages vector:
age_deciles <- quantile(ages, probs = decile_cumul_props)
age_deciles 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
20.170 25.585 32.750 38.040 42.830 47.710 52.170 56.210 60.750 64.290 69.750 Similarly, we can obtain the centiles as follows:
age_centiles <- quantile(ages, probs = seq(0, 1, 0.01))
age_centiles 0% 1% 2% 3% 4% 5% 6% 7% 8% 9%
20.1700 20.5935 21.0510 21.7060 22.2180 22.9800 23.3220 23.8345 24.1860 24.8300
10% 11% 12% 13% 14% 15% 16% 17% 18% 19%
25.5850 26.1700 26.8140 27.8300 28.8390 30.1025 30.5000 30.7500 31.5000 31.9625
20% 21% 22% 23% 24% 25% 26% 27% 28% 29%
32.7500 33.2620 33.7500 34.6485 35.5000 35.8300 36.2500 36.9200 37.4200 37.7780
30% 31% 32% 33% 34% 35% 36% 37% 38% 39%
38.0400 38.5800 38.8300 39.1700 39.8300 40.2500 40.6160 41.4640 41.7260 42.3945
40% 41% 42% 43% 44% 45% 46% 47% 48% 49%
42.8300 43.0935 43.7740 44.2060 44.4200 44.9200 45.5000 46.1700 46.7500 47.3300
50% 51% 52% 53% 54% 55% 56% 57% 58% 59%
47.7100 48.0800 48.4200 48.5800 49.0890 49.9200 50.3660 50.8795 51.1700 51.6565
60% 61% 62% 63% 64% 65% 66% 67% 68% 69%
52.1700 52.4320 52.8300 53.0000 53.3840 54.0400 54.5640 54.9240 55.4200 55.8300
70% 71% 72% 73% 74% 75% 76% 77% 78% 79%
56.2100 56.7220 57.1700 57.7460 58.0080 58.3525 59.0320 59.5440 59.9200 60.5000
80% 81% 82% 83% 84% 85% 86% 87% 88% 89%
60.7500 61.0935 61.5240 61.9200 62.2180 62.6700 63.0800 63.2540 63.6700 64.0800
90% 91% 92% 93% 94% 95% 96% 97% 98% 99%
64.2900 64.6700 65.1700 65.5760 65.8390 66.4200 66.7820 67.4235 68.1430 68.7245
100%
69.7500 7.5.5 Quantiles of theoretical distributions
R provides functions that give quantiles for certain common theoretical distributions.
These functions have a name starting with q and then something reminding the name of the theoretical distributions. For instance:
qunif, for the continuous uniform distribution;qnorm, for the normal distribution;qbinom, for the binomial distribution;qt, for the student distribution.
They all take as argument a vector containing probabilities.
They give back the corresponding quantiles, i.e. values where the cumulated probability reaches the corresponding probability.
These functions also take parameters of the distribution, if any. For instance, if we want the quantiles at probabilities \(0.05\), \(0.5\) and \(0.75\) for a normal distribution of mean \(2\) and standard deviation \(1.5\), we can use the mean and sd arguments of qnorm:
qnorm(c(0.05, 0.5, 0.75), mean = 2, sd = 1.5)[1] -0.4672804 2.0000000 3.0117346If a distribution is symmetric and centered on zero, the quantiles at \(p\) and \(1 - p\) have opposite values.
We can see this for instance for the standard normal distribution (by default, mean = 0 and sd = 1):
qnorm(0.05)[1] -1.644854qnorm(0.95)[1] 1.644854
The continuous uniform distribution models a random variable that can take any value within a given range of values, with equal probability.
For instance, here is the curve of the probability density for a distribution following the continous uniform model between \(0\) and \(2\):

What is its quantile at \(p = 0.75\) (that is, which value on the \(x\) axis has three quarters of the values below it, and one quarter above)?
7.6 Experimenting with random variables
Create a mean_5 vector containing the means of four series of 5 random numbers drawn from the standard normal distribution.
Now create a mean_100 vector containing the means of four series of of 100 random numbers drawn from the standard normal distribution.
Using sd, check that the means of many values are less dispersed that the means of a few values.

8 Matrices
Values can be organized in 2-dimensional structures called matrices.
8.1 Creating a Matrix
There are several ways to create matrices by taking data from vectors and organize it in two dimensions.
8.1.1 Reshaping a vector
One way to obtain a matrix is to give a vector to the matrix function, specifying the desired shape using the nrow and ncol arguments.
The vector will be used to fill the matrix column by column:
matrix(1:9, nrow = 3, ncol = 3) [,1] [,2] [,3]
[1,] 1 4 7
[2,] 2 5 8
[3,] 3 6 9It is possible to fill it row by row setting the byrow optional argument to TRUE:
matrix(1:9, nrow = 3, ncol = 3, byrow = TRUE) [,1] [,2] [,3]
[1,] 1 2 3
[2,] 4 5 6
[3,] 7 8 98.1.2 Stacking vectors together
Another way to create a matrix is to stack vectors horizontally (by binding rows with rbind) or vertically (by binding columns with cbind):
rbind(1:3, 4:6, 7:9) [,1] [,2] [,3]
[1,] 1 2 3
[2,] 4 5 6
[3,] 7 8 9cbind(1:3, 4:6, 7:9) [,1] [,2] [,3]
[1,] 1 4 7
[2,] 2 5 8
[3,] 3 6 98.2 Potential pitfalls when binding vectors
8.2.1 Type homogeneity and automatic conversion
Like vectors, matrices must contain values of the same type:
cbind(
1:3,
c(TRUE, FALSE, FALSE),
c("snow", "cloud", "vapour")) [,1] [,2] [,3]
[1,] "1" "TRUE" "snow"
[2,] "2" "FALSE" "cloud"
[3,] "3" "FALSE" "vapour" Notice how R automatically recoded our numbers and Booleans as character strings (without even warning us). That could be annoying, depending on what we planned to do with them. R automatically converting values from one type to another is a frequent cause for unexpected results.
We’ll see later that other structures exist in R, that can contain different types of values.
8.2.2 Element recycling
Before binding vectors into a matrix, you should ensure that their lengths are compatible. Indeed, R accepts combining vectors of different lengths, by “recycling” values as necessary:
cbind(1:4, c(1,2)) [,1] [,2]
[1,] 1 1
[2,] 2 2
[3,] 3 1
[4,] 4 2If the length of the longer vector is not a multiple of the lengths of the other ones, you will get a warning message:
cbind(1:5, c(1,2), c(3, 4, 5))Warning in cbind(1:5, c(1, 2), c(3, 4, 5)): number of rows of result is not a
multiple of vector length (arg 2) [,1] [,2] [,3]
[1,] 1 1 3
[2,] 2 2 4
[3,] 3 1 5
[4,] 4 2 3
[5,] 5 1 48.3 Transforming matrices
8.3.1 Growing
rbind and cbind can be used to add rows or columns to matrices.
Let’s create a square matrix containing 16 randomly generated normally distributed numbers:
We can do this starting with a vector of random numbers (obtained via the rnorm) function, and reshaping it with the matrix function:
random_16 <- matrix(rnorm(16), nrow = 4, ncol = 4)
random_16 [,1] [,2] [,3] [,4]
[1,] 1.1425763 2.4947919 0.5196763 -1.1393227
[2,] 0.2780924 -0.5739362 0.6074614 -0.8057326
[3,] 2.5973831 -0.6081713 0.1349975 -1.6214374
[4,] 0.4190047 1.0380363 0.7486941 -1.7437302Here is how we can add one more row, and then one more column:
random_20 <- rbind(random_16, rnorm(4))
random_20 [,1] [,2] [,3] [,4]
[1,] 1.1425763 2.494791917 0.5196763 -1.1393227
[2,] 0.2780924 -0.573936153 0.6074614 -0.8057326
[3,] 2.5973831 -0.608171302 0.1349975 -1.6214374
[4,] 0.4190047 1.038036340 0.7486941 -1.7437302
[5,] -0.1433717 -0.006419037 0.5708993 -0.3048804random_25 <- cbind(random_20, rnorm(5))
random_25 [,1] [,2] [,3] [,4] [,5]
[1,] 1.1425763 2.494791917 0.5196763 -1.1393227 -1.036718925
[2,] 0.2780924 -0.573936153 0.6074614 -0.8057326 0.005362493
[3,] 2.5973831 -0.608171302 0.1349975 -1.6214374 0.915051674
[4,] 0.4190047 1.038036340 0.7486941 -1.7437302 1.100020851
[5,] -0.1433717 -0.006419037 0.5708993 -0.3048804 -2.6886354398.3.2 Transposing
Sometimes, you may want to switch between two “orientations” of a matrix. You can apply this transposition operation, with the t function:
m1 <- rbind(1:3, 4:6, 7:9)
m1 [,1] [,2] [,3]
[1,] 1 2 3
[2,] 4 5 6
[3,] 7 8 9m2 <- t(m1)
m2 [,1] [,2] [,3]
[1,] 1 4 7
[2,] 2 5 8
[3,] 3 6 9We may guess from the very short name of this function that transposing matrices is expected to be a frequent operation, at least in the view of the designers of the language.
8.4 Matrix indexing
As suggested by the way the matrix is displayed, one can access the rows and columns they are made from, using square brackets ([]) with row or column indices.
my_matrix = cbind(1:3, 4:6, 7:9)
my_matrix [,1] [,2] [,3]
[1,] 1 4 7
[2,] 2 5 8
[3,] 3 6 9To access a row, put its index before a comma:
# second row
my_matrix[2,][1] 2 5 8 Don’t forget the comma, otherwise what you’ll get by writing my_matrix[2] is the “second” element of the matrix in a 1-dimensional indexing. Elements are counted from top to bottom, then left to right (the numbers in the current example do actually correspond to the element’s 1-dimensional indices).
To access a column, put its index after a comma:
# third column
my_matrix[,3][1] 7 8 9To access a single cell of a matrix, specify both indices (row, column):
# intersection of second row and third column
my_matrix[2,3][1] 8This is the same as extracting the second row (which is a vector), and then its third element:
# Same as:
# second_row <- my_matrix[2,]
# second_row[3]
my_matrix[2,][3][1] 8You can even specify slices:
my_matrix[,2:3] # equivalent to my_matrix[, c(2,3)] [,1] [,2]
[1,] 4 7
[2,] 5 8
[3,] 6 9Here, we extracted columns 2 to 3. As you can notice, the column indices are updated.
Now, can you think of a way to obtain all the columns, but in reverse order?
8.5 Operations on matrices
Like with vectors, certain operations can be applied on all elements of a matrix at once:
my_matrix + 1 [,1] [,2] [,3]
[1,] 2 5 8
[2,] 3 6 9
[3,] 4 7 10log(my_matrix, base = 2) [,1] [,2] [,3]
[1,] 0.000000 2.000000 2.807355
[2,] 1.000000 2.321928 3.000000
[3,] 1.584963 2.584963 3.169925my_matrix > 4 [,1] [,2] [,3]
[1,] FALSE FALSE TRUE
[2,] FALSE TRUE TRUE
[3,] FALSE TRUE TRUE# to the power 2 i.e. square
my_matrix ^ 2 [,1] [,2] [,3]
[1,] 1 16 49
[2,] 4 25 64
[3,] 9 36 81By the way, shortcut functions to compute logarithms of base 2 and 10 exist: log2 and log10.
9 INTERMISSION: Values can be classified into classes
In a sense, a matrix is a more complex kind of value than the basic kind of values that are vectors. Values can be classified into classes. One can get the class of a value using the class function:
class(c(1, 2, 3))[1] "numeric"class(c("a", "b"))[1] "character"class(FALSE)[1] "logical"We have already seen three basic sorts of values: numbers, character strings, and “logical” values (Booleans). What are the classes of our matrices?
class(my_matrix)[1] "matrix"class(matrix(c(FALSE, FALSE, FALSE, TRUE), nrow = 2, ncol = 2))[1] "matrix"Although we put different kinds of values inside (numbers in the first matrix, Booleans in the second), both are of the same kind of complex value (matrix), when considered as a whole.
We have seen another class, without mentioning it: tables.
class(table(mi$Sex))[1] "table"We will now learn about other classes: list and data.frame.
10 Lists
In R, a list is a way to group together different types of values.
10.1 Creating a list
You can create them with the list function:
gene_info <- list("WBGene00004775", "sep-1", "I", 3433156, 3438543, FALSE)
gene_info[[1]]
[1] "WBGene00004775"
[[2]]
[1] "sep-1"
[[3]]
[1] "I"
[[4]]
[1] 3433156
[[5]]
[1] 3438543
[[6]]
[1] FALSEclass(gene_info)[1] "list"The str function gives information about the structure of something in R:
str(gene_info)List of 6
$ : chr "WBGene00004775"
$ : chr "sep-1"
$ : chr "I"
$ : num 3433156
$ : num 3438543
$ : logi FALSEHere, we created a list containing six elements: three character strings, two numbers, and one Boolean (“logical” value).
10.2 Accessing list elements
As suggested by the way the list is displayed in the console, we can access the elements in the list using double square brackets and an index number:
gene_info[[2]][1] "sep-1"gene_info[[6]][1] FALSEIt can be useful to give names to the elements of the list.
This can be done using the names function:
names(gene_info) <- c("id", "name", "chrom", "start", "end", "fwd")
gene_info$id
[1] "WBGene00004775"
$name
[1] "sep-1"
$chrom
[1] "I"
$start
[1] 3433156
$end
[1] 3438543
$fwd
[1] FALSENotice the way the list is displayed now.
This suggests another way to access list elements:
gene_info$name[1] "sep-1"gene_info$fwd[1] FALSE(The double brackets way still works.)
You can also directly give names to list elements when you create it:
gene_info <- list(
id="WBGene00004775", name="sep-1",
chrom="I", start=3433156, end=3438543, fwd=FALSE)
gene_info$id
[1] "WBGene00004775"
$name
[1] "sep-1"
$chrom
[1] "I"
$start
[1] 3433156
$end
[1] 3438543
$fwd
[1] FALSE11 Data frames
Biological data usually consists in more complex information than what a type-homogeneous matrix can represent. Data points are characterized by various types of information: measurements (numbers), identifiers (strings or numbers), presence or absence of a treatment (Booleans), or discrete categories…
In R, data frames are commonly used for this kind of data. They consist in rows representing data points, and columns containing information about these data points.
They are a bit like a list (for the capability to hold different types of elements), and a bit like a matrix (for the organization in rows and columns). Some of the things you learned with matrices and lists will apply also to data frames. That’s actually the reason why we included matrices and lists in the course.
11.1 Creating data frames
You can build a data frame by joining vectors, using the data.frame function, where each vector becomes a column representing an element of information about a series of data points (all vectors must therefore have the same length):
# Here, we wrote the arguments on several lines for clarity.
data.frame(
1:5,
c(FALSE, FALSE, TRUE, FALSE, FALSE),
c("cow", "ewe", "cow", "goat", "soy"),
c(4, 5, 3, 3, 1))| X1.5 | c.FALSE..FALSE..TRUE..FALSE..FALSE. | c..cow….ewe….cow….goat….soy.. | c.4..5..3..3..1. |
|---|---|---|---|
| 1 | FALSE | cow | 4 |
| 2 | FALSE | ewe | 5 |
| 3 | TRUE | cow | 3 |
| 4 | FALSE | goat | 3 |
| 5 | FALSE | soy | 1 |
The columns got very ugly default header names. Let’s save a cleaner data frame with more explicit column names in a cheese variable. You can directly specify the names of the columns by naming your vectors in the data.frame function call (just like we did for list items):
cheese <- data.frame(
id=1:5,
cooked=c(FALSE, FALSE, TRUE, FALSE, FALSE),
milk=c("cow", "ewe", "cow", "goat", "soy"),
strength=c(4, 5, 3, 3, 1))
class(cheese) # Just to be sure[1] "data.frame"cheese| id | cooked | milk | strength |
|---|---|---|---|
| 1 | FALSE | cow | 4 |
| 2 | FALSE | ewe | 5 |
| 3 | TRUE | cow | 3 |
| 4 | FALSE | goat | 3 |
| 5 | FALSE | soy | 1 |
(It is also possible to change the column names in an existing data frame via the names function (as for lists and named vectors), or colnames, which is more explicit.)
We can even specify row names to make more explicit what our data points represent, using the function rownames:
rownames(cheese) <- c(
"Camembert", "Roquefort", "Comté",
"Pélardon", "Tofu")
cheese| id | cooked | milk | strength | |
|---|---|---|---|---|
| Camembert | 1 | FALSE | cow | 4 |
| Roquefort | 2 | FALSE | ewe | 5 |
| Comté | 3 | TRUE | cow | 3 |
| Pélardon | 4 | FALSE | goat | 3 |
| Tofu | 5 | FALSE | soy | 1 |
(Don’t you dare doubting about the scientificity and accuracy of my cheese strength scale!)
11.2 Indexing in data frames
We can extract part of a data frame as we did with matrices, using numeric indices:
# first row
cheese[1,]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Camembert | 1 | FALSE | cow | 4 |
Vectors of indices can also be used:
# columns 2 to 3
cheese[,2:3]| cooked | milk | |
|---|---|---|
| Camembert | FALSE | cow |
| Roquefort | FALSE | ewe |
| Comté | TRUE | cow |
| Pélardon | FALSE | goat |
| Tofu | FALSE | soy |
And we can extract single cells:
cheese[2,2][1] FALSEHeader and row names can be used as well for sub-setting:
cheese["Comté",]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Comté | 3 | TRUE | cow | 3 |
cheese[,"cooked"] # or, like with lists cheese$cooked[1] FALSE FALSE TRUE FALSE FALSE How would you check if Tofu is made from cooked paste?
11.3 Boolean sub-setting
Like with vectors, row sub-setting can be done with vectors of Booleans. The selection vector should be put before a comma, to indicate that rows are being selected (not columns):
cheese[cheese$cooked,]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Comté | 3 | TRUE | cow | 3 |
The above syntax works by selecting those rows for which the corresponding value in the selection vector is TRUE.
Indeed, the cooked column is a vector of Booleans:
is_cooked <- cheese$cooked
is_cooked[1] FALSE FALSE TRUE FALSE FALSEIt can be used to select rows:
cheese[is_cooked,]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Comté | 3 | TRUE | cow | 3 |
cheese[cheese$strength > 3,]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Camembert | 1 | FALSE | cow | 4 |
| Roquefort | 2 | FALSE | ewe | 5 |
Similarly, comparing a vector of numbers with another number results in a vector of Booleans:
is_strong <- cheese$strength > 3
is_strong[1] TRUE TRUE FALSE FALSE FALSEHence:
cheese[is_strong,]| id | cooked | milk | strength | |
|---|---|---|---|---|
| Camembert | 1 | FALSE | cow | 4 |
| Roquefort | 2 | FALSE | ewe | 5 |
Now extract those cheese that are not made of cow’s milk.
11.4 Adding columns
New columns can be created and added to an existing data frame. This can be done easily by assigning a vector to a non existing column.
Try to add a column is_strong with Boolean values indicating whether the cheese is considered strong (strength at least 4) or not.
11.5 Modeling discrete categories using factors
If we look at the structure of our data frame, we’ll notice that it’s milk column is actually not made of character strings:
str(cheese)'data.frame': 5 obs. of 5 variables:
$ id : int 1 2 3 4 5
$ cooked : logi FALSE FALSE TRUE FALSE FALSE
$ milk : Factor w/ 4 levels "cow","ewe","goat",..: 1 2 1 3 4
$ strength : num 4 5 3 3 1
$ is_strong: logi TRUE TRUE FALSE FALSE FALSEBy default data.frame converts character strings into factors.
Factors are a way to describe discrete categories. They are actually yet another class of data:
class(cheese$milk)[1] "factor"11.5.1 Avoiding factors
Could we have avoided the automatic conversion when creating our cheese data frame? Let’s find out using the help!
?data.frameThere’s a stringsAsFactors option in data.frame. Let’s try it to create another version of the data frame where the milk column is kept as strings:
cheese2 <- data.frame(
id=1:5,
cooked=c(FALSE, FALSE, TRUE, FALSE, FALSE),
milk=c("cow", "ewe", "cow", "goat", "soy"),
strength=c(4, 5, 3, 3, 1),
stringsAsFactors=FALSE)
str(cheese2)'data.frame': 5 obs. of 4 variables:
$ id : int 1 2 3 4 5
$ cooked : logi FALSE FALSE TRUE FALSE FALSE
$ milk : chr "cow" "ewe" "cow" "goat" ...
$ strength: num 4 5 3 3 1class(cheese2$milk)[1] "character"11.5.2 What if we want factors?
If we are in a case where factors are useful, we can use conversion from strings to factors with the as.factor function:
milks <- as.factor(c("cow", "ewe", "cow", "goat", "soy"))
milks[1] cow ewe cow goat soy
Levels: cow ewe goat soyThe levels represent the categories. Here, we have four such categories.
Levels can be displayed using the levels function:
levels(milks)[1] "cow" "ewe" "goat" "soy" We can also use levels to add new levels to our vector of factors, without adding elements:
levels(milks) <- c(levels(milks), "poppy")If we count the number of elements for each category using table, we will also get counts for the levels having no instances in our factors:
table(milks)milks
cow ewe goat soy poppy
2 1 1 1 0 The tapply function allows us to apply another function separately on categories. For instance, if we want to compute separate mean strengths for each type of milk, we can proceed as follows:
tapply(cheese$strength, cheese$milk, mean) cow ewe goat soy
3.5 5.0 3.0 1.0 (This will actually work also if the second argument can be converted into factors.)


11.6 Importing your own data
So far, we have worked with a data frame that we created from within R. In real life, you will much more often work with data present in files.
When working with files in R, it is important to know where the files are with respect to the location in your computer that the R session uses as a reference, its working directory.
11.6.1 Working directory
Files and directories in a computer are located using paths, which are represented as strings.
Let’s display the path of our current working directory using the function getwd (“get working directory”):
getwd()[1] "/builds/hub-courses/R_pasteur_phd"(By the way, RStudio shows the current working directory at the top of the “Console” tab.)
We are going to create a new directory with dir.create:
dir_name <- "stat_course"
dir.create(dir_name)Then set this new directory as our current working directory, specifying its path with the function setwd:
setwd('stat_course') # or setwd(dir_name)
# And finally verify that the change took place:
getwd()[1] "/builds/hub-courses/R_pasteur_phd/stat_course"From now on, if we try to read or write files from R, this will take place relatively to the new current working directory.
In RStudio, you can also change the working directory via the “Session” > “Set Working Directory” > “Choose Directory” menu.
It is very important to “know where we are” in order to find our files and not create a mess or even loose important data by accidentally overwriting files.
11.6.2 Downloading files
We provide files containing counts for a sample of genes in RNA-seq data libraries:
WT_1: https://c3bi.pasteur.fr/wp-content/uploads/2018/09/WT_1.tsvWT_2: https://c3bi.pasteur.fr/wp-content/uploads/2018/09/WT_2.tsvKO_1: https://c3bi.pasteur.fr/wp-content/uploads/2018/09/KO_1.tsvKO_2: https://c3bi.pasteur.fr/wp-content/uploads/2018/09/KO_2.tsv
(The data is also present in the full course archive.)
Each file is in plain text format, and consists in two columns separated by a tabulation. Here are the first lines of one of them:
gene_name WT_1
sep-1 8702 # Be careful with gene names that Excel may convert into a date.
tag-63 3470
msh-6 9444
egg-4 3002
egg-5 3379
tofu-4 1026
klp-15 5989
trpp-8 3931
cir-1 3064You can see that the first line contains column headers. The first column contains gene names. The second contains the corresponding read counts for library WT_1 (first replicate of a wild type).
Create a data directory, download the datasets and save them in this directory.
Try to open one of those files with your favorite spreadsheet program. It should be possible. Have a look at the first gene name, has it been converted to a date (see https://doi.org/10.1186/s13059-016-1044-7)?
11.6.3 Loading data from a file
Now let’s do this in R. First verify that the previously downloaded data folder is present in your working directory:
list.files()The data can be loaded into a data frame using read.table:
counts_WT_1 <- read.table(
"data/WT_1.tsv",
header=TRUE,
sep="\t",
stringsAsFactors=FALSE)read.table can take many arguments. Here are those we just used:
- The
headerargument is set toTRUEto indicate that the first line of the file has to be used to define the column names. - The
separgument specifies what column separator should be used to correctly interpret the data. Remember that"\t"is a special notation to represent a tabulation. - By default, columns of character strings are recoded into factors when R builds a data frame. It is better to define factors when we really need them instead of letting R do it for us. For now, we want to keep the information as character strings, using
stringsAsFactors=FALSE.
11.6.4 Data inspection
It is a good idea to have a quick look at the imported data before doing some real work with it. Let’s check the first rows with the head function:
head(counts_WT_1)| gene_name | WT_1 |
|---|---|
| sep-1 | 8702 |
| tag-63 | 3470 |
| msh-6 | 9444 |
| egg-4 | 3002 |
| egg-5 | 3379 |
| tofu-4 | 1026 |
There is also a tail function, that extracts the last rows.
To know how many rows and columns our data frame contains, we can use the nrow and ncol functions:
nrow(counts_WT_1)[1] 196ncol(counts_WT_1)[1] 2This data frame has 196 rows and 2 columns.
We can use the dim (dimension) function to directly get those two pieces of information:
dim(counts_WT_1)[1] 196 2More details can be obtained about the structure of the data frame, using str:
str(counts_WT_1)'data.frame': 196 obs. of 2 variables:
$ gene_name: chr "sep-1" "tag-63" "msh-6" "egg-4" ...
$ WT_1 : int 8702 3470 9444 3002 3379 1026 5989 3931 3064 5245 ...This tells us that counts_WT_1 is a data.frame with 196 rows and 2 columns:
gene_name, which contains character strings (chr);WT_1, which contains integral numbers (int).
We can also use the summary function, that we already tried on a vector:
summary(counts_WT_1) gene_name WT_1
Length:196 Min. : 1
Class :character 1st Qu.: 1310
Mode :character Median : 3969
Mean : 6309
3rd Qu.: 6942
Max. :107158 What we get here is a summary for each column. Depending on the type of content of the column, the information given by summary differs.
12 Data frame use case: Finding genes impacted by a mutation
The four provided datasets correspond to a sample of gene counts in RNA-seq data in a wild type (WT) nematode (C. elegans) and a mutant (KO) of some gene expression regulation pathway. There are two replicates for each condition (_1 and _2).
We want to find what genes are the most up-regulated or down-regulated in the mutant. To do this, we will proceed through the following steps:
- (Obviously,) load the data
- Merge count data by first joining replicates for each condition (and check that replicates are similar enough), then joining the conditions in a single data frame
- Normalize the counts
- Compute mean normalized counts for each condition
- Compute the fold change: the ratio of the mean normalized counts for the mutant over that for the wild type.
- Find the genes having the maximum and minimum fold change.
12.1 Loading the data
First, load the four datasets in four data frames (counts_WT_1, counts_WT_2, counts_KO_1 and counts_KO_2). How would you do that?
Let’s have a quick look at the data:
str(counts_WT_1)'data.frame': 196 obs. of 2 variables:
$ gene_name: chr "sep-1" "tag-63" "msh-6" "egg-4" ...
$ WT_1 : int 8702 3470 9444 3002 3379 1026 5989 3931 3064 5245 ...str(counts_WT_2)'data.frame': 196 obs. of 2 variables:
$ gene_name: chr "sep-1" "tag-63" "msh-6" "egg-4" ...
$ WT_2 : int 7973 3204 8478 2840 3028 904 5401 3810 2849 5019 ...str(counts_KO_1)'data.frame': 192 obs. of 2 variables:
$ gene_name: chr "sep-1" "tag-63" "msh-6" "egg-4" ...
$ KO_1 : int 6183 2570 6456 1827 1806 731 3679 3836 2267 3817 ...str(counts_KO_2)'data.frame': 194 obs. of 2 variables:
$ gene_name: chr "sep-1" "tag-63" "msh-6" "egg-4" ...
$ KO_2 : int 5776 2273 6198 1474 1431 682 2966 3892 2167 3484 ...Note that they do not all have the same number of rows. One consequence is that we will have to deal with missing data.
Another consequence is that a given row index (line number) does not necessarily correspond to the same gene in different data frames. (This could be true even with the same number of rows.)
12.2 Merging data frames
12.2.1 Joining replicates
We will start by merging counts of replicates in a common data frame. This is a job for the merge function:
counts_WT <- merge(counts_WT_1, counts_WT_2, by = "gene_name", all = FALSE)
counts_KO <- merge(counts_KO_1, counts_KO_2, by = "gene_name", all = FALSE)Here are the arguments that we used:
- The first two arguments are the data frames to merge.
- The
byargument indicates on the basis of which column those data frames should be merged. - The
allargument indicates whether we want to keep all rows (addingNAwhere values are missing), or only those that exist in both input data frames. Here, we reason that data absent from one replicate must not be very reliable, so we setall = FALSE.
Other potentially useful arguments are available (see ?merge).
A quick look shows that we now have data frames with one gene_name column, and one column per replicate:
head(counts_WT)| gene_name | WT_1 | WT_2 |
|---|---|---|
| abcf-1 | 6431 | 5984 |
| acdh-13 | 2141 | 1957 |
| adr-1 | 3498 | 3236 |
| alh-9 | 4249 | 3346 |
| ari-1 | 2464 | 2301 |
| arp-1 | 3153 | 2985 |
head(counts_KO)| gene_name | KO_1 | KO_2 |
|---|---|---|
| abcf-1 | 5395 | 5655 |
| acdh-13 | 1913 | 1815 |
| adr-1 | 2697 | 2782 |
| alh-9 | 3792 | 4014 |
| ari-1 | 2073 | 2071 |
| arp-1 | 2811 | 2955 |
Notice that the rows have been sorted by gene name. We could have avoided that by using the sort = FALSE argument of merge.
12.2.1.1 The scatter plot
It is a good idea to check the correlation between replicates, so let’s compare both groups of replicates by plotting counts of one against the other:
plot(x = counts_WT$WT_1, y = counts_WT$WT_2)
plot(x = counts_KO$KO_1, y = counts_KO$KO_2)
This representation is called a scatter plot. Each dot represents one gene. The coordinates of a point on the \(x\) and \(y\) axes represent the counts for the corresponding genes in the replicates.
The lowest values are not well visible, log transforming the data enhances differences in these values:
plot(x = log(counts_WT$WT_1), y = log(counts_WT$WT_2))
plot(x = log(counts_KO$KO_1), y = log(counts_KO$KO_2))
12.2.1.2 Correlation coefficients
Besides checking graphically, it is possible to describe the correlation between paired series of values using correlation coefficients. One of them is Pearson’s correlation coefficient. Usually noted \(r\), it is a value between \(-1\) and \(1\).
When \(r\) is close to \(1\), the values in the two series are positively correlated (when a high value is observed in one series, the value for the corresponding observation in the other series tends to be high too) and the points in the scatter plot tend to be aligned along a line with a positive slope. When \(r\) is close to \(-1\), the values are negatively correlated, and the corresponding point also tend to be aligned, but along a line with a negative slope. When \(r\) is close to \(0\), the values are not correlated, and the corresponding dots are far from being aligned.
Pearson’s \(r\) can be obtained using the cor function:
cor(counts_WT$WT_1, counts_WT$WT_2)[1] 0.9992361cor(counts_KO$KO_1, counts_KO$KO_2)[1] 0.9978398 Generate two vectors of \(50\) random values drawn from standard normal distributions, plot their scatterplot and compute their correlation coefficient.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
You should be aware that Pearson’s \(r\) is sensitive to the presence of outliers.
Other kinds of correlation coefficients exist, such as Spearman’s correlation rank correlation coefficient, usually noted \(\rho\).
12.2.2 Joining conditions
Now that you know how to join data frames together, create a counts_all data frame with the two we just created.
Have a look at the structure of this new data frame. How many rows do we have now?
We can now start to compute serious stuff, like normalized values and fold changes.
12.3 Normalizing the counts
In a given sample, the number of read counts for a given gene likely depends on the total number of read counts.
12.3.1 Computing the total number of read counts
What’s the total counts for each sample?
We can see that the wild type samples have a noticeably larger number of counts than the mutant ones. To have comparable values between samples, we will compute the proportion of total counts that a gene represents.
12.3.2 Creating tables of normalized counts
Create a new data frame from norm_all with five columns:
- the original
gene_namecolumn; - a column obtained by dividing the columns containing the counts by the corresponding sum of counts, for each sample.
12.4 Computing mean normalized counts
The rowMeans function, as its name suggests, computes the mean of each row in a data frame.
We can use this to compute the means for the two conditions, and add new columns to our data frame:
norm_all$WT_mean <- rowMeans(norm_all[, c("WT_1", "WT_2")])
norm_all$KO_mean <- rowMeans(norm_all[, c("KO_1", "KO_2")]) Similar functions exist to work on columns or to compute sums instead: colMeans, rowSums, colSums.
12.5 Computing fold changes
Using the techniques seen in the previous sections, you will now add a fold_change column containing the KO_mean / WT_mean ratios for each gene.
(We want values higher than 1 for genes that are up-regulated in the KO.)
12.6 Finding the most affected genes
Our work will now consist in the following steps:
- Computing the log fold changes.
- Adding a column with a status indication depending on the log fold change value.
- Sorting the table by log fold change.
- Writing the results in a file.
12.6.1 Computing the log fold change
Log fold change (LFC) is a commonly used measure of change in gene expression level.
For instance, a gene with a 4-fold increase in the KO with respect to the wild type will have a LFC value of 2, and a gene with a 4-fold decrease will have a LFC of -2.
Use the log2 function to add a LFC column indicating the base 2 logarithm of the fold change.
12.6.2 Indicating gene regulation status
For the sake of the exercise, let’s decide that we consider “strongly up-regulated” a gene with more than than a 2-fold increase in the KO, and “strongly down-regulated” a gene with more than a 2-fold decrease.
Using ifelse, add a status column, with either "up", "down" or "." (for the genes not strongly affected).
What are the strongly up-regulated genes?
And the strongly down-regulated?
It seems that histone genes (name starting with "his-") are particularly affected in this experiment.
How can we extract a data frame containing only the information regarding histone genes? (Save the result in a new norm_hist data frame.)
Now, let’s visualize the distribution of statuses among the histone genes:
barplot(table(norm_hist$status))
And among the whole data set:
barplot(table(norm_all$status))
12.6.3 Sorting the data
To sort a vector we can use the order function, which generates the order of the indices we should use in order to select the elements in sorted order:
my_numbers <- c(5, 1, 3, 2)
# increasing order, by default (should be 2, 4, 3, 1)
order(my_numbers)[1] 2 4 3 1my_numbers[order(my_numbers)][1] 1 2 3 5# decreasing order (should be 1, 3, 4, 2)
order(my_numbers, decreasing = TRUE)[1] 1 3 4 2my_numbers[order(my_numbers, decreasing = TRUE)][1] 5 3 2 1We can do the same with the LFC column:
order(norm_all$LFC, decreasing = TRUE) [1] 80 104 37 189 121 81 12 4 41 102 179 103 131 40 162 126 23 6
[19] 139 133 84 8 122 2 138 33 83 178 140 157 125 118 18 1 169 113
[37] 154 5 116 119 172 111 127 44 149 136 145 9 142 32 22 180 156 21
[55] 187 3 165 101 107 146 161 185 155 164 20 15 13 173 170 160 14 124
[73] 35 123 153 17 34 64 77 168 11 120 177 152 166 39 148 171 159 175
[91] 163 31 158 25 181 24 132 114 128 105 30 190 184 134 7 106 129 182
[109] 45 147 99 19 110 26 112 117 137 135 115 141 183 10 42 176 93 174
[127] 167 66 151 43 38 130 144 48 186 108 90 55 100 27 16 109 86 95
[145] 143 89 29 91 188 28 79 57 87 75 94 76 96 85 150 36 88 54
[163] 60 63 92 82 62 74 65 69 61 46 56 67 97 68 70 78 58 59
[181] 72 47 51 71 98 53 50 73 52 49 We can now use this to select the rows by their indices, and we should obtain a table where the rows are ordered by decreasing LFC values.
Do it, and name the resulting table sorted_on_LFC:
12.6.4 Saving the results in a file
Be careful when writing files: No warnings will be shown if the file already exists, so you may accidentally overwrite precious data.
Therefore, before saving anything in a file, check with list.files() that the file name you are planning to use does not correspond to an existing file.
A while ago, you learned how to read a table from a file using read.table. Guess what the write.table function does?
Look at the help of this function and use it to save the sorted data frame in a file called KO_vs_WT_LFC.tsv within your current working directory. We want the columns to be separated by tabulations, and we don’t want character strings to be written with double quotes around them.
13 More practice with Milieu Intérieur
13.1 Reminder: Loading the data
Milieu Intérieur survey data mi.csv can be obtained here: http://hub-courses.pages.pasteur.fr/R_pasteur_phd/data/mi.csv
Download and load the dataset into your R session (the mi.csv file should be in your data folder):
(The data is also present in the full course archive.)
This file is in the plain text format CSV for Comma-Separated values. Columns are here separated by commas and not tabulation like in TSV-formatted files.
The mi.csv data frame contains a header and a column with subject ids. We specify this extra information to properly import the data frame.
mi_data = read.table("data/mi.csv", sep = ",", header = TRUE, row.names = "SUBJID")13.2 Descriptive statistics
To get an idea of the shape of the data frame, we can use dim:
dim(mi_data)[1] 816 43Or its structure can be examined with str:
str(mi_data)'data.frame': 816 obs. of 43 variables:
$ X : int 1 2 3 4 5 6 7 8 9 10 ...
$ Age : num 22.3 28.8 23.7 21.2 26.2 ...
$ OwnsHouse : Factor w/ 2 levels "No","Yes": 2 2 2 1 2 2 1 2 1 2 ...
$ PhysicalActivity : num 3 0 0 0.5 1.5 0 4 0 0 1.5 ...
$ Sex : Factor w/ 2 levels "Female","Male": 1 1 1 1 1 1 1 1 1 1 ...
$ LivesWithPartner : Factor w/ 2 levels "No","Yes": 1 2 2 1 1 2 2 1 2 1 ...
$ LivesWithKids : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ BornInCity : Factor w/ 2 levels "No","Yes": 2 2 2 1 2 2 2 2 2 1 ...
$ Inbreeding : num 95 79.1 117.3 94.2 105.1 ...
$ BMI : num 20.1 21.3 22.2 18.7 29 ...
$ CMVPositiveSerology : Factor w/ 2 levels "No","Yes": 1 2 1 1 1 1 1 2 1 1 ...
$ FluIgG : num 0.4643 -0.0498 0.3329 0.4049 -0.3038 ...
$ MetabolicScore : int 0 1 2 0 1 1 1 0 1 1 ...
$ LowAppetite : int 0 0 0 0 0 3 0 3 0 0 ...
$ TroubleConcentrating: int 0 0 0 0 0 0 0 0 0 0 ...
$ TroubleSleeping : num 1 1 1 2 1 1 0 0 1 3 ...
$ HoursOfSleep : num 9 7.05 6.5 10 9 8 8 7 7 7 ...
$ Listless : int 3 3 3 3 0 3 0 3 0 3 ...
$ UsesCannabis : Factor w/ 2 levels "No","Yes": 1 1 2 1 1 1 2 2 1 2 ...
$ RecentPersonalCrisis: Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 2 1 1 ...
$ Smoking : Factor w/ 3 levels "Active","Ex",..: 3 1 1 3 3 3 1 1 3 3 ...
$ Employed : Factor w/ 2 levels "No","Yes": 1 2 2 1 2 2 2 2 2 1 ...
$ Education : Factor w/ 5 levels "Baccalaureat",..: 3 1 1 3 1 1 5 1 1 3 ...
$ DustExposure : Factor w/ 3 levels "Current","No",..: 2 2 1 2 2 2 3 2 2 2 ...
$ Income : Factor w/ 4 levels "(1000-2000]",..: 1 2 2 3 4 1 1 1 2 1 ...
$ HadMeasles : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 2 1 1 1 ...
$ HadRubella : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ HadChickenPox : Factor w/ 2 levels "No","Yes": 2 2 2 2 1 2 2 2 2 1 ...
$ HadMumps : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 2 1 1 1 ...
$ HadTonsillectomy : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ HadAppendicectomy : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ VaccineHepA : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ VaccineMMR : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 2 1 1 ...
$ VaccineTyphoid : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ VaccineWhoopingCough: Factor w/ 2 levels "No","Yes": 2 2 1 1 2 2 1 2 1 1 ...
$ VaccineYellowFever : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 1 1 ...
$ VaccineHepB : Factor w/ 2 levels "No","Yes": 2 2 2 2 2 1 2 2 2 2 ...
$ VaccineFlu : Factor w/ 2 levels "No","Yes": 1 1 1 1 1 1 1 1 2 1 ...
$ DepressionScore : num 0 0 0 1 0 0 0 2 0 1 ...
$ HeartRate : int 66 66 62 64 67 72 61 71 53 58 ...
$ Temperature : num 36.8 37.4 36.9 36 36.7 36.6 36.8 36.9 36.7 36.8 ...
$ HourOfSampling : num 8.88 9.35 8.67 9.88 8.55 ...
$ DayOfSampling : int 40 40 40 40 81 81 82 82 109 110 ...To list the variables considered, we can display de name of the columns:
names(mi_data) [1] "X" "Age" "OwnsHouse"
[4] "PhysicalActivity" "Sex" "LivesWithPartner"
[7] "LivesWithKids" "BornInCity" "Inbreeding"
[10] "BMI" "CMVPositiveSerology" "FluIgG"
[13] "MetabolicScore" "LowAppetite" "TroubleConcentrating"
[16] "TroubleSleeping" "HoursOfSleep" "Listless"
[19] "UsesCannabis" "RecentPersonalCrisis" "Smoking"
[22] "Employed" "Education" "DustExposure"
[25] "Income" "HadMeasles" "HadRubella"
[28] "HadChickenPox" "HadMumps" "HadTonsillectomy"
[31] "HadAppendicectomy" "VaccineHepA" "VaccineMMR"
[34] "VaccineTyphoid" "VaccineWhoopingCough" "VaccineYellowFever"
[37] "VaccineHepB" "VaccineFlu" "DepressionScore"
[40] "HeartRate" "Temperature" "HourOfSampling"
[43] "DayOfSampling" Calculate the median and mean of the Age:
median(mi_data$Age)[1] 47.71mean(mi_data$Age)[1] 46.48571We can plot for example the distribution of temperature by gender:
plot(mi_data$Temperature~mi_data$Sex)
We can also obtain a summary:
summary(mi_data) X Age OwnsHouse PhysicalActivity Sex
Min. : 1.0 Min. :20.17 No :528 Min. : 0.000 Female:417
1st Qu.:204.8 1st Qu.:35.83 Yes:288 1st Qu.: 0.500 Male :399
Median :408.5 Median :47.71 Median : 2.000
Mean :408.5 Mean :46.49 Mean : 2.752
3rd Qu.:612.2 3rd Qu.:58.35 3rd Qu.: 4.000
Max. :816.0 Max. :69.75 Max. :49.000
LivesWithPartner LivesWithKids BornInCity Inbreeding BMI
No :315 No :458 No :382 Min. : 43.73 Min. :18.50
Yes:501 Yes:358 Yes:434 1st Qu.: 84.08 1st Qu.:21.77
Median : 91.86 Median :23.85
Mean : 91.90 Mean :24.21
3rd Qu.:100.01 3rd Qu.:26.21
Max. :150.11 Max. :32.00
CMVPositiveSerology FluIgG MetabolicScore LowAppetite
No :527 Min. :-0.43049 Min. :0.0000 Min. : 0.0000
Yes:289 1st Qu.: 0.06508 1st Qu.:0.0000 1st Qu.: 0.0000
Median : 0.22785 Median :1.0000 Median : 0.0000
Mean : 0.20360 Mean :0.9326 Mean : 0.5123
3rd Qu.: 0.36382 3rd Qu.:1.0000 3rd Qu.: 0.0000
Max. : 0.76984 Max. :4.0000 Max. :14.0000
TroubleConcentrating TroubleSleeping HoursOfSleep Listless
Min. : 0.0000 Min. :0.00 Min. : 3.000 Min. : 0.00
1st Qu.: 0.0000 1st Qu.:0.00 1st Qu.: 7.000 1st Qu.: 0.00
Median : 0.0000 Median :1.00 Median : 7.500 Median : 0.00
Mean : 0.3554 Mean :1.12 Mean : 7.499 Mean : 1.29
3rd Qu.: 0.0000 3rd Qu.:2.00 3rd Qu.: 8.000 3rd Qu.: 3.00
Max. :14.0000 Max. :3.00 Max. :12.000 Max. :14.00
UsesCannabis RecentPersonalCrisis Smoking Employed Education
No :769 No :580 Active:161 No :395 Baccalaureat:219
Yes: 47 Yes:236 Ex :223 Yes:421 Bachelor :120
Never :432 PhD :125
UpToPrimary : 73
Vocational :279
DustExposure Income HadMeasles HadRubella HadChickenPox HadMumps
Current: 68 (1000-2000]:262 No :504 No :740 No :294 No :585
No :607 (2000-3000]:230 Yes:312 Yes: 76 Yes:522 Yes:231
Past :141 (3000-inf] :220
[0-1000] :104
HadTonsillectomy HadAppendicectomy VaccineHepA VaccineMMR VaccineTyphoid
No :750 No :628 No :780 No :647 No :775
Yes: 66 Yes:188 Yes: 36 Yes:169 Yes: 41
VaccineWhoopingCough VaccineYellowFever VaccineHepB VaccineFlu
No :616 No :748 No :413 No :655
Yes:200 Yes: 68 Yes:403 Yes:161
DepressionScore HeartRate Temperature HourOfSampling
Min. : 0.0000 Min. : 37.00 Min. :35.70 Min. : 8.433
1st Qu.: 0.0000 1st Qu.: 54.00 1st Qu.:36.20 1st Qu.: 8.917
Median : 0.0000 Median : 58.00 Median :36.40 Median : 9.233
Mean : 0.5445 Mean : 59.21 Mean :36.43 Mean : 9.215
3rd Qu.: 1.0000 3rd Qu.: 65.00 3rd Qu.:36.60 3rd Qu.: 9.550
Max. :14.0000 Max. :100.00 Max. :37.70 Max. :11.217
DayOfSampling
Min. : 17.0
1st Qu.:136.0
Median :187.0
Mean :185.5
3rd Qu.:263.0
Max. :335.0 14 Problem solving
It is perfectly normal to experience difficulties such as code that doesn’t work, or doesn’t work as expected. This happens everyday to advanced practitioners, so why not to beginners too?
This chapter tries to gather some pieces of advice regarding how to overcome such difficulties.
14.1 Checking the documentation
Remember that R has an internal help system accessible from the interactive console.
The help of R functions is not necessarily easy to understand, but it should be quick and cheap to have a look at it, just to be sure that you use the arguments as expected, and what the default values are for the arguments you don’t set.
You can also have a look at resources given in the References.
14.2 Experimenting
Some of us are more comfortable with experimenting than with delving in the documentation.
Experimenting with the R language is a perfectly legitimate way to acquire an understanding of its way of working. Debugging a program can be done using a trial-and-error approach. So don’t hesitate to test things, in the R console, for a start.
14.3 Asking questions properly
If, after some reasonable efforts, you don’t come up with a solution for your problem, you may ask others for help.
These pages will help you maximizing your chances of getting help from the internet people:
14.4 Stack exchange sites
If you use the right words, you may find answers on these great “Question and Answers” websites:
- About R: https://stackoverflow.com/questions/tagged/r
- About statistics: https://stats.stackexchange.com/
First try to search for existing answers. Often, others have had the same problem as you, and may have obtained relevant answers. If your search fails, ask questions (keeping in mind the advice learned from the previous section).
14.5 Rocket.Chat
Institut Pasteur has an internal chat system.
You can make contact with local R users using the following channel: https://rocketchat.pasteur.cloud/channel/r
This is of course only accessible to people having an account on the intranet of Institut Pasteur.
14.6 The Bioinformatics and Biostatistics Hub
The engineers of the Hub may provide you some assistance if you work in a Pasteur Institute. The two main possibilities are the following:
Open desks: If you work in Institut Pasteur in Paris, you can come and see us on Tuesdays from 10 to 12 in the hall of the Yersin building (building number 24).
Short questions: https://c3bi.pasteur.fr/services/ask-question/
15 Practice
Please, do not forget to go through the few following exercises before the next session: http://hub05.hosting.pasteur.fr/klebsiella_ace/training/prelim.Rmd
They will provide you some important elements to get started with “Confidence Intervals & Statistical Testing” module, but can be useful for other modules in our biostatistics and bioinformatics courses, and more generally to make sure you have acquired some of the most essential R skills from the present course.
(If you plan to follow other courses, we also recommend you to check whether you can install some packages in advance.)
16 References
16.1 The classics
16.2 MOOC on edx
16.3 In French
http://www.info.univ-angers.fr/~gh/tuteurs/statistiques_pour_statophobes.pdf
https://pascalneige.files.wordpress.com/2011/09/r-pour-les-statophobes.pdf
Reference book by our colleague Gaël Millot (restricted access): Comprendre et réaliser les tests statistiques à l’aide de R
17 In case of temporary boredom
- Look at the optional sections.
- Learn more about functions (see below)
- Read the help.
- Learn Python and Julia too!
17.1 Experimenting with built-in datasets
We have already briefly seen how to create graphics. Plotting data is actually one of the points where R is powerful.
R provides datasets readily available to play with and test your plotting skills:
data()To load the “iris” dataset:
data("iris")An iris data frame is now available.
str(iris)'data.frame': 150 obs. of 5 variables:
$ Sepal.Length: num 5.1 4.9 4.7 4.6 5 5.4 4.6 5 4.4 4.9 ...
$ Sepal.Width : num 3.5 3 3.2 3.1 3.6 3.9 3.4 3.4 2.9 3.1 ...
$ Petal.Length: num 1.4 1.4 1.3 1.5 1.4 1.7 1.4 1.5 1.4 1.5 ...
$ Petal.Width : num 0.2 0.2 0.2 0.2 0.2 0.4 0.3 0.2 0.2 0.1 ...
$ Species : Factor w/ 3 levels "setosa","versicolor",..: 1 1 1 1 1 1 1 1 1 1 ...We can plot for example the distribution of sepal lengths by species:
plot(iris$Sepal.Length~iris$Species)
The Y~X syntax is called a formula. It is used to express a dependency, a relationship, between variables. The ~ (“tilda”) sign roughly means “as a function of”. Here, we are asking a plot of the relationship between sepal length and species.
17.2 Factoring code with functions
In programming, it is important to avoid repetitions (according to the DRY principle). A function is a way to group together a set of instructions and call them at will. Functions can take zero or more arguments, which are meant to influence the function’s outcome.
17.2.1 My first homemade function
Tradition suggests we start by writing a function that prints a simple and friendly message:
helloWorld <- function() cat("Hello, world!")The function we just defined consists in just one instruction: executing the cat function with a predefined string as argument.
We save it using the variable name helloWorld, and then call it by using parentheses after its name.
helloWorld()Hello, world!This is just like a (short)cut to print “Hello, world!”, and may not seem very useful.
Now imagine that you wrote a script that prints “Hello, world!” hundreds of times. You change your mind, and want your script to print “That’s all, folks!” instead. If you used a function for that, you will just need to modify the message once (in the function definition) instead of doing it hundreds of times. Remember that computers should help you with repetitive tasks: they are really good at it if you use them the right way.
17.2.2 Functions with arguments
To define functions that can adapt their behaviour depending on some values given as argument, put argument names inside the parentheses of the function definition:
howAreYouFr <- function(name) cat("Ça va", name, "?")
howAreYouFr("Manu")Ça va Manu ?The value given inside the parentheses when calling the function will be associated to the corresponding argument name. This results in a variable that can be used inside the function (here, as an argument to cat).
Such variables are not visible outside of the function:
name## Error: object 'name' not found17.2.3 Functions with multiple instructions
A function can consist in multiple instructions. In that case, you need to indicate where instructions belonging to the function start and end, using curly braces to delimit the {function body}.
echo <- function(sentence) {
cat(sentence)
cat("\n") # Otherwise nothing separates the sentences
# We could also have added a new line character to the sentence:
# cat(paste(sentence, "\n", sep = ""))
cat(sentence)
}
echo("Hiho!")Hiho!
Hiho!The function body can be written like any normal code, and can include comments.
Here, we wrote the function body “indented” to try to improve readability by making the function body more easily distinguishable from the rest of the code. When typing in the console, you get those confusing + signs, but if you write your function in a script, it really helps.
You can also write everything in one line, separating instructions with semi-colons (;):
echo2 <- function(sentence) {cat(paste0(sentence, "\n")); cat(sentence)}
echo2("Hoooo!")Hoooo!
Hoooo!This style is more adapted for simple and short functions that you want to quickly define while working in the interactive console.
17.2.4 Functions with a result
The last instruction to be evaluated in a function, when it results in a value, can be used in further computations:
squareDiagonal <- function(side1, side2) {
square1 = side1 * side1
square2 = side2 * side2
# According to Pythagoras:
square1 + square2
}
diagonal <- sqrt(squareDiagonal(3, 4))
diagonal[1] 5It is common to say that such a function returns a value.
Remember when we learned how to take decisions using Booleans?
We can now create a function that simply encapsulates our work with ifelse:
verdict <- function(grade) ifelse(grade >= 10, "passed", "failed")When we want to give a verdict, we just have to call our verdict function:
verdict(4)[1] "failed"verdict(17)[1] "passed"verdict(c(6, 18, 14))[1] "failed" "passed" "passed"17.2.5 Variable scope
Variables can be defined or modified inside the function body without affecting variables already existing outside: Variables have different scopes.
To study what is happening with variables inside and outside of a function, we will create one that prints its internal variables. Here we will use the print function, because it is more convenient than cat for this type of experiments. It doesn’t display special characters such as \n or \t nicely, but as they appear in the console, returning to a new line afterwards.
x <- 1
variablePrinter <- function(v) {
print("argument v:")
print(v)
v <- v * 3
print("modified v:")
print(v)
x <- v - 1
}
y <- variablePrinter(x)[1] "argument v:"
[1] 1
[1] "modified v:"
[1] 3Inside the function, v took the value that we gave as argument, that is, the value of x (1). Still inside the function, we modify v, tripling its value to 3
Adding print instructions in the code to obtain informations about your variables is a common “quick-and-dirty”" technique to debug programs.
What is the value of y?
What is the value of x in the global scope?
And what about v?
17.2.6 Applying functions on multiple values
Some functions having one argument can be applied to vectors with multiple elements.
square <- function(number) number * number
square(1:10) [1] 1 4 9 16 25 36 49 64 81 100This results in a vector where each value of the original one has been used to produce a new value.
Of course, this is a very simple example, and one could simply do 1:10 * 1:10. This technique can be useful with more complex cases.
17.2.7 Further functional programming
Some functions take functions as their arguments.
For instance, sapply allows you to apply a function (given as second argument) to every value of a vector (given as first argument):
squares <- sapply(c(1, 2, 3), function(number) number * number)Note that a function can be defined on the spot, directly as an argument to another function. If we don’t need it outside of the function, we don’t have to associate it to a variable name beforehand.
squares[1] 1 4 9The result is a vector, consisting in as many elements as there were in the one given to sapply. It is equivalent to write c(1 * 1, 2 * 2, 3 * 3).
It seems simpler to define the square function and give it directly a vector as argument, as we did before. However, for some specific functions, this may not work as intended. sapply is more robust.
One can apply functions that take multiple arguments to multiple vectors of arguments, using mapply:
mapply(squareDiagonal, c(1, 3, 6), c(1, 4, 8))[1] 2 25 100Here, the result is the same as writing c(squareDiagonal(1, 1), squareDiagonal(3, 4), squareDiagonal(6, 8))
17.3 Packages
A package is a way to obtain functionalities not present in the default version of R. Generally, a package depends on other packages, called its dependencies. These packages can be used to extend built-in R functionalities.
If you follow other modules in our biostatistics and bioinformatics courses, you will need to install some packages.
We have been informed of the following package requirements by other modules:
| Multivariate analysis | Functional analysis |
|---|---|
| FactoMineR | AnnotationDbi |
| RColorBrewer | DESeq2 |
| cluster | FactoMineR |
| factoextra | KEGGREST |
| corrplot | RColorBrewer |
| gplots | dplyr |
| ggplot2 | factoextra |
| ggplot2 | |
| limma | |
| org.Hs.eg.db | |
| pheatmap | |
| png | |
| progress |
If you plan to follow one of these modules, please try to install the corresponding packages in advance.
Note that some of them have to be installed using Bioconductor which will be discussed in a later section.
17.3.1 Installing packages using the RStudio graphical interface
In R studio, go to the “Packages” tab (by default, in the bottom-right pane) and click “Install”. You can then search for the package you want to install and install it along its dependencies (check the box “Install dependencies”).
17.3.2 Installing packages using the console
To install a package, you can also simply use its name as argument to the install.packages function. For example to install the dplyr package:
install.packages("dplyr")What can you try right now in order to avoid wasting time installing packages during the “Multivariate analysis” course?
17.3.3 Troubleshooting installation
If the installation fails, you may have to install other things which are not part of R, like compilers (tools that transform code into working executable programs) and development libraries (files providing more or less general functionalities that multiple programs can use).
Watch out for possible error messages during the installation process. This will give you ideas of keywords to use when searching for a solution on the internet in case something goes wrong.
If you are using Mac OSX, you might find some of the missing tools here: https://cran.rstudio.com/bin/macosx/tools/.
If you are using Linux, the information here might help: https://github.com/blaiseli/R_install_notes/blob/master/Install_Linux.md.
Sometimes, a failure to install packages may be due to incompatibilities between packages. In such a case, something you can try is to use an alternate package manager, such as Bioconductor.
17.3.4 Bioconductor
There is an effort of the bioinformatics community to provide a set of specialized R packages through it’s own system: Bioconductor (which actually uses install.packages under the hood).
Bioconductor strives to provide a set of compatible packages, and can also install R packages that are not specific to bioinformatics.
To be able to install packages via Bioconductor, you should first install the BiocManager package:
install.packages("BiocManager")Then, you can install packages using the BiocManager::install function. For instance, to install the differential expression analysis package DESeq2:
BiocManager::install("DESeq2")You will likely be able to use this instead of install.packages in most cases, not only for bioinformatics-specific tools.
For instance if you encountered issues installing packages for PCA you may try the following:
BiocManager::install("FactoMineR")
BiocManager::install("factoextra")17.3.5 Using a package
Once the package of interest is installed, we should be able to use functions provided by this package.
To load all the functions of the package into your current session, you can use the function library: For example with the dplyr package:
library("dplyr")
# Then the "full_join" function from dplyr is available for the current session:
counts_WT_bis <- full_join(counts_WT_1, counts_WT_2, by = "gene_name")Or you can use a specific function from this package using the package name and double colons (::) as prefix.
For example, if you want to use only the function full_join of the dplyr package:
counts_WT_bis <- dplyr::full_join(counts_WT_1, counts_WT_2, by = "gene_name")18 Cheat Sheet
18.1 Number notation
.: decimal separator in a number2e3:2times (10to the power of3)1L: forced integral number
18.2 Operators
+,-,*,/: the standard mathematical operators%%,%/%: modulus and integer divisionx^n:xto the power ofnlog2(x), log10(x), log(x, base=n): logarithms<,>,<=,>=,==,!=: comparison operators
18.3 Boolean computations
TRUE,FALSE: literal Boolean values (without quotes)|,&,!: Boolean OR, AND and NOT.ifelse(condition, do_if_true, do_otherwise): deciding what to do depending on the Booleanconditionis.na: test whether values areNA(== NAreturnsNA)
18.4 Strings
"word": literal character strings are between quotes"": empty stringpaste(s1, s2, sep=s): join stringss1ands2using separators(s1ands2can also be vectors of strings)"\t","\n": special characters for tabulation and new linecat: human-friendly displaying of strings
18.5 Factors and formulae
stringsAsFactors=FALSE: tell some functions to keep strings as stringsas.factor(s): convert stringsinto factorslevels(f): get or set the levels of factorsffactor(c("a", "b"), levels=c("a", "b", "c")): factors with extra levelstapply(v, f, func): applyfuncon elements ofvpartitioned according to the corresponding factors inf- formulae?
18.6 Vector creation
c: combine values of a same type in a vector (c()creates an empty vector)c(a=1, b=2): create a vector with named elementsn:m: Vector of numbers fromntomseq(n, m, by=i): sequence fromntomincrementing byirep: repeat valuesrnorm(n, mean=m, sd=s):nrandom numbers from a normal distributionsample(v, n, replace=TRUE): random sample ofnelements from vectorv, with possibly multiple occurrences of an elementvector(length=n, mode="numeric")initialize a vector withndefault numeric values (0)
18.7 Repeating a computation
replicate(n, f(x)):nvalues obtained by callingf(x)for (i in 1:n) {f(i)}: repeat a computation withisuccessively assuming the values from1:ni <- 1; while (i <= n) {f(i); i <- i + 1}: repeat a computation while a condition isTRUE(;\(\Leftrightarrow\) new line)
18.8 Operations on vectors
- values get recycled when operating between vectors
length(v): number of elements invv[n]: get then\(^{th}\) value inv(also works with multiple indices)v[n:m]: get from then\(^{th}\) to them\(^{th}\) value invv[b], withba vector of Booleans: get the values fromvwherebisTRUEwhich[b]: indices wherebisTRUEsum(v): sum of the elements inv
18.9 Matrices
cbind,rbind: stack vectors by columns or by rows (recycling shorter vectors and converting types if necessary)matrix(v, nrow=n, ncol=m): reshape vectorvinto ann\(\times\)mmatrixdim: get or set number of rows and columnsmat[n,]: getn\(^{th}\) row (several rows iflength(n) > 1)mat[,m]: getm\(^{th}\) column (several rows iflength(m) > 1)mat[n,m]: get value at coordinates(n, m)t(mat): transpose matrixmat
18.10 Lists
list(A=v1, B=v2): list with two named elementsAandBmade from arbitrary valuesv1andv2(names are optional)names: get or set elements namesl[[n]]: get then\(^{th}\) elementl$A: get the element namedA
18.11 Data frames
data.frame(A=c1, B=c2): data frame with two named columnsAandBmade from vectorsv1andv2(names are optional)data.frame(..., stringsAsFactors=FALSE): keep strings as stringsnames,colnames: get or set column namesrownames: get or set row namesdf[n,]: getn\(^{th}\) row (several rows iflength(n) > 1) as a data frame (row names can be used instead of numbers)df[b,]: get rows where Boolean vectorbisTRUE, as a data framedf$A,df[,"A"],df[,m]: get columnA(orm\(^{th}\)) as a vectordf[, c("A", "B")],df[, 1:2]: get columnsAandB(or from1to2) as a data frame (more than one column cannot be just a vector)df[n,m]: get value at rownand columnm(numbers or row and column names can be used)df$C <- v3: change or add columnCusing vectorv3merge(df1, df2, by="A"): merge data framesd1andd2based on columnArowMeans,rowSums,colMeans,colSums: quick means or sums of rows or cols (argumentna.rm=TRUEto ignore missing values)
18.12 Paths, files, input / output
getwd: Where am I?setwd(path): I want to be atpath.dir.create(path): creating a directory atpathcat(string, file=path): write string into file atpathread.table(path): read file atpathinto data frame, various arguments:header=TRUE: use first line as column namesstringsAsFactors=FALSE: keep strings as stringssep="\t": column separator is tabulation
write.table(df, file=path): write data framedfinto file atpath, various arguments:sep="\t": column separator is tabulation
18.13 Functions
f <- function(args) {body}: defining a functionfusingargsin codebodyf(args): executingfusingargsfunction() {instr_1; instr_2}: return the value ofinstr_2(;\(\Leftrightarrow\) new line)function() {return instr_1; instr_2}: return the value ofinstr_1, don’t executeinstr_2
18.14 Examining, visualizing
head,tail: first and last elementsnrow,ncol,dim: get dimensionsmin,max,range,mean,var,sd,median,quantile,IQR,summary: descriptive statisticsclass: information about class of datastr: structure of somethingtable: easy countinghist: plot histogrambarplot: plot bar plotsboxplot: box-and-whiskers plotsplot: versatile plotting functionabline: straight lines on an existing plotpar: graphical parameters (to customize your graphs)