4 Data Types¶
4.1 Exercices¶
4.1.1 Exercise¶
Assume that we execute the following assignment statements:
width = 17
height = 12.0
delimiter ='.'
For each of the following expressions, write the value of the expression and the type (of the value of the expression) and explain.
- width / 2
- width / 2.0
- height / 3
- 1 + 2 * 5
Use the Python interpreter to check your answers.
>>> width = 17
>>> height = 12.0
>>> delimiter ='.'
>>>
>>> width / 2
8
>>> # both operands are integer so python done an euclidian division and threw out the remainder
>>> width / 2.0
8.5
>>> height / 3
4.0
>>> # one of the operand is a float (2.0 or height) then python pyhton perform afloat division but keep in mind that float numbers are aproximation.
>>> # if you need precision you need to use Decimal. But operations on Decimal are slow and float offer quite enought precision
>>> # so we use decimal only if wee need great precision
>>> # Euclidian division
>>> 2 / 3
0
>>> # float division
>>> float(2)/float(3)
0.6666666666666666
>>> # decimal division
>>> from decimal import Decimal
>>> a = Decimal(2)
>>> b = Decimal(3)
>>> a / b
Decimal('0.6666666666666666666666666667')
>>> 1 + 2 * 5
11
4.1.2 Exercise¶
Write a function which take a radius as input and return the volume of a sphere:
The volume of a sphere with radius r is 4/3 πr3.
What is the volume of a sphere with radius 5?
Hint: π is in math module, so to access it you need to import the math module
Place the import statement at the top fo your file.
after that, you can use math.pi everywhere in the file like this:
>>> import math
>>>
>>> #do what you need to do
>>> math.pi #use math.pi
Hint: the volume of a spher with radius 5 is not 392.7 !
1 2 3 4 5 6 7 8 9 | import math
def vol_of_sphere(radius):
"""
compute the volume of sphere of a given radius
"""
vol = float(4)/float(3) * float(math.pi) * pow(radius, 3)
return vol
|
python -i volume_of_sphere.py
>>> vol_of_sphere(5)
523.5987755982989
4.1.3 Exercise¶
Draw what happen in memory when the following statements are executed:
i = 12
i += 2
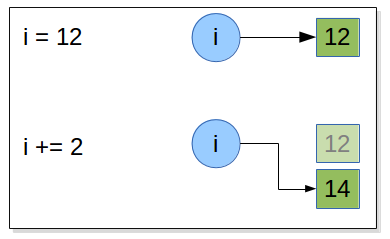
>>> i = 12
>>> id(i)
33157200
>>> i += 2
>>> id(i)
33157152
and
s = 'gaa'
s = s + 'ttc'
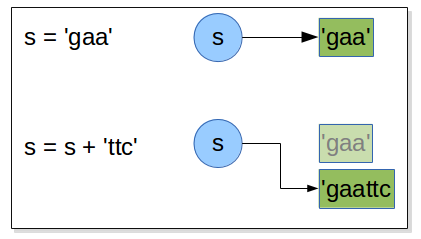
>>> s = 'gaa'
>>> id(s)
139950507582368
>>> s = s+ 'ttc'
>>> s
'gaattc'
>>> id(s)
139950571818896
when an augmented assignment operator is used on an immutable object is that
- the operation is performed,
- and an object holding the result is created
- and then the target object reference is re-bound to refer to the result object rather than the object it referred to before.
So, in the preceding case when the statement i += 2 is encountered, Python computes 1 + 2 , stores
the result in a new int object, and then rebinds i to refer to this new int . And
if the original object a was referring to has no more object references referring
to it, it will be scheduled for garbage collection. The same mechanism is done with all immutable object included strings.
4.1.4 Exercise¶
- how to obtain a new sequence which is the 10 times repetition of the this motif : “AGGTCGACCAGATTANTCCG”::
>>> s = "AGGTCGACCAGATTANTCCG" >>> s10 = s * 10
4.1.5 Exercise¶
create a representation in fasta format of following sequence :
Note
A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The description line is distinguished from the sequence data by a greater-than (“>”) symbol in the first column. The word following the “>” symbol is the identifier of the sequence, and the rest of the line is the description (optional). There should be no space between the “>” and the first letter of the identifier. The sequence ends if another line starting with a “>” appears; this indicates the start of another sequence.
id = "sp|P60568|IL2_HUMAN"
comment = "Interleukin-2 OS=Homo sapiens GN=IL2 PE=1 SV=1"
sequence = """MYRMQLLSCIALSLALVTNSAPTSSSTKKTQLQLEHLLLDLQMILNGINNYKNPKLTRML
TFKFYMPKKATELKHLQCLEEELKPLEEVLNLAQSKNFHLRPRDLISNINVIVLELKGSE
TTFMCEYADETATIVEFLNRWITFCQSIISTLT"""
>>> s = id + comment + '\n' + sequence
or
>>> s = "{id} {comment} \n{sequence}".format(id= id, comment = comment, sequence = sequence)
4.1.6 Exercise¶
For the following exercise use the python file sv40 in fasta which is a python file with the sequence of sv40 in fasta format
already embeded, and use python -i sv40_file.py to work.
how long is the sv40 in bp? Hint : the fasta header is 61bp long. (http://www.ncbi.nlm.nih.gov/nuccore/J02400.1)
pseudocode
write a function fasta_to_one_line that return a sequence as a string
without header or any non sequence characters
pseudocode:
1 2 3 4 5 6 7 |
def fasta_to_one_line(seq):
header_end_at = seq.find('\n')
seq = seq[header_end_at + 1:]
seq = seq.replace('\n', '')
return seq
|
python
>>> import sv40_file
>>> import fasta_to_one_line
>>>
>>> sv40_seq = fasta_to_one_line(sv40_file.sv40_fasta)
>>> print(len(sv40_seq))
5243
Is that the following enzymes:
- BamHI (ggatcc),
- EcorI (gaattc),
- HindIII (aagctt),
- SmaI (cccggg)
have recogition sites in sv40 (just answer by True or False)?
>>> "ggatcc".upper() in sv40_sequence
True
>>> "gaattc".upper() in sv40_sequence
True
>>> "aagctt".upper() in sv40_sequence
True
>>> "cccggg".upper() in sv40_sequence
False
for the enzymes which have a recognition site can you give their positions?
>>> sv40_sequence = sv40_sequence.lower()
>>> sv40_sequence.find("ggatcc")
2532
>>> # remind the string are numbered from 0
>>> 2532 + 1 = 2533
>>> # the recognition motif of BamHI start at 2533
>>> sv40_sequence.find("gaattc")
1781
>>> # EcorI -> 1782
>>> sv40_sequence.find("aagctt")
1045
>>> # HindIII -> 1046
is there only one site in sv40 per enzyme?
The find method give the index of the first occurence or -1 if the substring is not found.
So we can not determine the occurence of a site only with the find method.
We will see how to do that when we learn looping and conditions.
4.1.7 Exercise¶
We want to perform a PCR on sv40, can you give the length and the sequence of the amplicon?
Write a function which have 3 parameters sequence, primer_1 and primer_2
- We consider only the cases where primer_1 and primer_2 are present in sequence
- to simplify the exercise, the 2 primers can be read directly on the sv40 sequence.
test you algorithm with the following primers
Write the pseudocode before to implement it.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 |
def amplicon_len(seq, primer_1, primer_2):
"""
given primer_1 and primer_2
return the lenght of the amplicon
primer_1 and primer_2 ar present and in this order
in sequence
"""
primer_1 = primer_1.upper()
primer_2 = primer_2.upper()
sequence = sequence.upper()
pos_1 = sv40_sequence.find(primer_1)
pos_2 = sv40_sequence.find(primer_2)
amplicon_len = pos_2 + len(primer_2) - pos_1
return amplicon_len
|
>>> import sv40
>>> import fasta_to_one_line
>>>
>>> sequence = fasta_to_one_line(sv40)
>>> print(amplicon_len(sequence, first_primer, second_primer))
199
4.1.8 Exercise¶
reverse the following sequence “TACCTTCTGAGGCGGAAAGA” (don’t compute the complement):
>>> "TACCTTCTGAGGCGGAAAGA"[::-1]
or
>>> s = "TACCTTCTGAGGCGGAAAGA"
>>> l = list(s)
# take care reverse() reverse a list in place (the method do a side effect and return None )
# so if you don't have a object reference on the list you cannot get the reversed list!
>>> l.reverse()
>>> print(l)
>>> ''.join(l)
or
>>> rev_s = reversed(s)
''.join(rev_s)
The most efficient way to reverse a string or a list is the way using the slice.
4.1.9 Exercise¶
We have to take care of the string numbered vs sequence numbered:
il2_human = ‘MYRMQLLSCIALSLALVTNSAPTSSSTKKTQLQLEHLLLDLQMILNGINNYKNPKLTRMLTFKFYMPKKATELKHLQCLEEELKPLEEVLNLAQSKNFHLRPRDLISNINVIVLELKGSETTFMCEYADETATIVEFLNRWITFCQSIISTLT’
head = il2_human[:77]
body = il2_human[77:125]
tail = il2_human[126:]
body_mutate = body.replace('C', 'S')
il2_mutate = head + body_mutate + tail
4.1.10 Exercise¶
Write a function
- which take a sequence as parameter
- compute the GC%
- and return it
- display the results readable for human as a micro report like this: ‘the sv40 is 5243 bp length and have 40.80% gc’
use sv40 sequence to test your function.
1 2 3 4 |
def gc_percent(seq):
gc_pc = float(seq.count('g') + seq.count('c')) / float(len(seq))
return gc_pc
|
>>> import sv40
>>> import fasta_to_one_line
>>> import gc_percent
>>>
>>> sequence = fasta_to_one_line(sv40)
>>> gc_pc = gc_percent(sequence)
>>> report = "the sv40 is {0} bp length and have {1:.2%} gc".format(len(sequence), gc_pc)
>>> print report
'the sv40 is 5243 bp length and have 40.80% gc'
gc_pc = float(sv40_sequence.count('g') + sv40_sequence.count('c')) / float(len(sv40_sequence))
"the sv40 is {0} bp lenght and have {1:.2%} gc".format(len(sv40), gc_pc)