13 Object Oriented Programming¶
13.1 Exercises¶
13.1.1 Exercise¶
Modelize a sequence with few attributes and methods
1class Sequence(object):
2
3 def __init__(self, identifier, comment, seq):
4 self.id = identifier
5 self.comment = comment
6 self.seq = self._clean(seq)
7
8
9 def _clean(self, seq):
10 """
11 remove newline from the string representing the sequence
12 :param seq: the string to clean
13 :return: the string without '\n'
14 :rtype: string
15 """
16 return seq.replace('\n')
17
18
19 def gc_percent(self):
20 """
21 :return: the gc ratio
22 :rtype: float
23 """
24 seq = self.seq.upper()
25 return float(seq.count('G') + seq.count('C')) / len(seq)
26
27
28
29
30dna1 = Sequence('gi214', 'the first sequence', 'tcgcgcaacgtcgcctacatctcaagattca')
31dna2 = Sequence('gi3421', 'the second sequence', 'gagcatgagcggaattctgcatagcgcaagaatgcggc')
13.1.2 Exercise¶
Instanciate 2 sequences using your Sequence class, and draw schema representing the namespaces
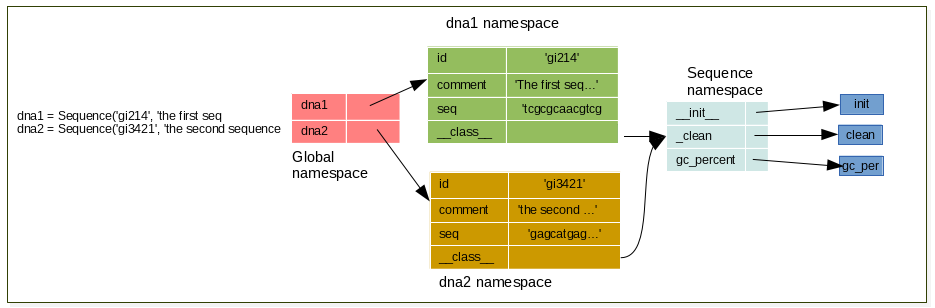

13.1.3 Exercise¶
Can you explain this result (draw namespaces to explain) ? how to modify the class variable class_attr
1class MyClass(object):
2
3 class_attr = 'foo'
4
5 def __init__(self, val):
6 self.inst_attr = val
7
8
9
10
11a = MyClass(1)
12b = MyClass(2)
13
14print a.inst_attr
151
16print b.inst_attr
172
18
19print a.class_attr == b.class_attr
20True
21print a.class_attr is b.class_attr
22True
23
24b.class_attr = 4
25
26print a.class_attr
274
28del a.class_attr
29
30MyClass.class_attr = 4
13.1.4 Exercise¶
Write the definition of a Point class. Objects from this class should have a
a method show to display the coordinates of the point
a method move to change these coordinates.
a method dist that computes the distance between 2 points.
Note
the distance between 2 points A(x0, y0) and B(x1, y1) can be compute
(http://www.mathwarehouse.com/algebra/distance_formula/index.php)
The following python code provides an example of the expected behaviour of objects belonging to this class:
>>> p1 = Point(2, 3)
>>> p2 = Point(3, 3)
>>> p1.show()
(2, 3)
>>> p2.show()
(3, 3)
>>> p1.move(10, -10)
>>> p1.show()
(12, -7)
>>> p2.show()
(3, 3)
>>> p1.dist(p2)
1.0
1import math
2
3
4class Point(object):
5 """Class to handle point in a 2 dimensions space"""
6
7 def __init__(self, x, y):
8 """
9 :param x: the value on the X-axis
10 :type x: float
11 :param y: the value on the Y-axis
12 :type y: float
13 """
14 self.x = x
15 self.y = y
16
17
18 def show(self):
19 """
20 :return: the coordinate of this point
21 :rtype: a tuple of 2 elements (float, float)
22 """
23 return (self.x, self.y)
24
25
26 def move(self, x, y):
27 """
28 :param x: the value to move on the X-axis
29 :type x: float
30 :param y: the value to move on the Y-axis
31 :type y: float
32 """
33 self.x += x
34 self.y += y
35
36
37 def dist(self, pt):
38 """
39 :param pt: the point to compute the distance with
40 :type pt: :class:`Point` object
41 :return: the distance between this point ant pt
42 :rtype: int
43 """
44 dx = pt.x - self.x
45 dy = pt.y - self.y
46 return math.sqrt(dx ** 2 + dy ** 2)
point.py .
13.1.5 Exercise¶
Use biopython to read a fasta file (sv40.fasta)
and display the attributes
id
name
description
seq
use the module SeqIO in biopython A tutorial is available https://biopython.org/wiki/SeqIO
from Bio import SeqIO
sv40_rcd = SeqIO.read("sv40.fasta", "fasta")
print("id =", sv40_rcd.id)
print("name =", sv40_rcd.name)
print("description =", sv40_rcd.description)
print("sequence =", sv40_rcd.seq)
Other example of usage of SeqIO: seq_io.py
13.1.6 Exercise¶
Translate the sequence in phase 1, 2, -2
sv40_seq_phase1 = sv40_rcd.seq
sv40_seq_phase2 = sv40_rcd[1:]
sv40_seq_phase_2 = sv40_rcd[1:].reverse_complement(id=True)
13.1.7 Exercise¶
Create a sequence with the first 42 nucleotides
Translate this sequence
Mutate the nucleotide in position 18 ‘A’ -> ‘C’
and translate the mutated sequence
see tutorial http://biopython.org/DIST/docs/tutorial/Tutorial.html#htoc28
short_seq = sv40_seq_phase2[0:42]
short_seq.translate()
mutable_seq = short_seq.seq.tomutable()
mutable_seq[19] = 'C'
mutate_seq = mutable_seq.toseq()
mutate_seq.translate()
13.1.8 Exercise¶
Open the file abcd.fasta (abcd.fasta) and convert it in genbank format
Hint: the seq alphabet attribute must be set to extended_protein see Bio.Alphabet.IUPAC module
from Bio.Alphabet.IUPAC import extended_protein
with open("abcd.fasta", "r") as fasta, open('abcd.gb', 'w') as genbank:
for record in SeqIO.parse(fasta, "fasta"):
record.seq.alphabet = extended_protein
print(len(record.seq))
SeqIO.write(record, genbank, 'genbank')
13.1.9 Exercice¶
Open the file abcd.fasta (abcd.fasta) and filter out sequence <= 700
Write the results in fasta file
with open("abcd.fasta", "r") as input, open("abcd_short.fasta", "w") as output:
for record in SeqIO.parse(input, "fasta"):
if len(record.seq) > 700:
SeqIO.write(record, output, 'fasta')
13.1.10 Exercise¶
Use OOP to modelize restriction enzyme, and sequences.
the sequence must implement the following methods
enzyme_filter which take as a list of enzymes as argument and return a new list containing the enzymes which have binding site in sequence
the restriction enzyme must implements the following methods
binds which take a sequence as argument and return True if the sequence contains a binding site, False otherwise.
solve the exercise 7.1.4 Exercise using this new implementation.
1
2class Sequence(object):
3
4 def __init__(self, identifier, comment, seq):
5 self.id = identifier
6 self.comment = comment
7 self.seq = self._clean(seq)
8
9
10 def _clean(self, seq):
11 """
12
13 :param seq:
14 :return:
15 """
16 return seq.replace('\n')
17
18 def enzyme_filter(self, enzymes):
19 """
20
21 :param enzymes:
22 :return:
23 """
24 enzymes_which_binds = []
25 for enz in enzymes:
26 if enz.binds(self.seq):
27 enzymes_which_binds.append(enz)
28 return
29
30
31class RestrictionEnzyme(object):
32
33 def __init__(self, name, binding, cut, end, comment=''):
34 self._name = name
35 self._binding = binding
36 self._cut = cut
37 self._end = end
38 self._comment = comment
39
40 @property
41 def name(self):
42 return self._name
43
44 def binds(self, seq):
45 """
46
47 :param seq:
48 :return:
49 """
50 return self.binding in seq.seq
13.1.11 Exercise¶
refactor your code of 8.1.15 Exercise in OOP style programming. implements only
size: return the number of rows, and number of columns
get_cell: that take the number of rows, the number of columns as parameters, and returns the content of cell corresponding to row number col number
set_cell: that take the number of rows, the number of columns as parameters, and a value and set the value val in cell specified by row number x column number
to_str: return a string representation of the matrix
mult: that take a scalar and return a new matrix which is the scalar product of matrix x val
you can change the name of the methods to be more pythonic
1
2
3
4
5class Matrix(object):
6
7 def __init__(self, row, col, val=None):
8 self._row = row
9 self._col = col
10 self._matrix = []
11 for i in range(row):
12 c = [val] * col
13 self._matrix.append(c)
14
15 def size(self):
16 return self._row, self._col
17
18 def get_cell(self, row, col):
19 self._check_index(row, col)
20 return self._matrix[i][j]
21
22 def matrix_set(self, row, col, val):
23 self._check_index(row, col)
24 self._matrix[row][col] = val
25
26 def __str__(self):
27 s = ''
28 for i in range(self._row):
29 s += self._matrix[i]
30 s += '\n'
31 return s
32
33 def _check_index(self, row, col):
34 if not (0 < row <= self._row) or not (0 < col <= self._col):
35 raise IndexError("matrix index out of range")
13.1.12 Exercise¶
Use the code to read multiple sequences fasta file in procedural style and refactor it in OOP style.
use the file abcd.fasta to test your code.
What is the benefit to use oop style instead of procedural style?
1class Sequence(object):
2
3 def __init__(self, id_, sequence, comment=''):
4 self.id = id_
5 self.comment = comment
6 self.sequence = sequence
7
8 def gc_percent(self):
9 seq = self.sequence.upper()
10 return float(seq.count('G') + seq.count('C')) / float(len(seq))
11
12class FastaParser(object):
13
14
15 def __init__(self, fasta_path):
16 self.path = fasta_path
17 self._file = open(fasta_path)
18 self._current_id = ''
19 self._current_comment = ''
20 self._current_sequence = ''
21
22 def _parse_header(self, line):
23 """
24 parse the header line and _current_id|comment|sequence attributes
25 :param line: the line of header in fasta format
26 :type line: string
27 """
28 header = line.split()
29 self._current_id = header[0][1:]
30 self._current_comment = ' '.join(header[1:])
31 self._current_sequence = ''
32
33 def __iter__(self):
34 return self
35
36 def next(self):
37 """
38 :return: at each call return a new :class:`Sequence` object
39 :raise: StopIteration
40 """
41 for line in self._file:
42 if line.startswith('>'):
43 # a new sequence begin
44 if self._current_id != '':
45 new_seq = Sequence(self._current_id,
46 self._current_sequence,
47 comment=self._current_comment)
48 self._parse_header(line)
49 return new_seq
50 else:
51 self._parse_header(line)
52 else:
53 self._current_sequence += line.strip()
54 if not self._current_id and not self._current_sequence:
55 self._file.close()
56 raise StopIteration()
57 else:
58 new_seq = Sequence(self._current_id,
59 self._current_sequence,
60 comment=self._current_comment)
61 self._current_id = ''
62 self._current_sequence = ''
63 return new_seq
64
65
66if __name__ == '__main__':
67 import sys
68 import os.path
69
70 if len(sys.argv) != 2:
71 sys.exit("usage fasta_object fasta_path")
72 fasta_path = sys.argv[1]
73 if not os.path.exists(fasta_path):
74 sys.exit("No such file: {}".format(fasta_path))
75
76 fasta_parser = FastaParser(fasta_path)
77 for sequence in fasta_parser:
78 print "----------------"
79 print "{seqid} = {gc:.3%}".format(gc=sequence.gc_percent(),
80 seqid = sequence.id)