 TC, BN, JBM, AZ
TC, BN, JBM, AZ
Institut Pasteur, Paris, 20-31 March 2017
Overview of data structures¶
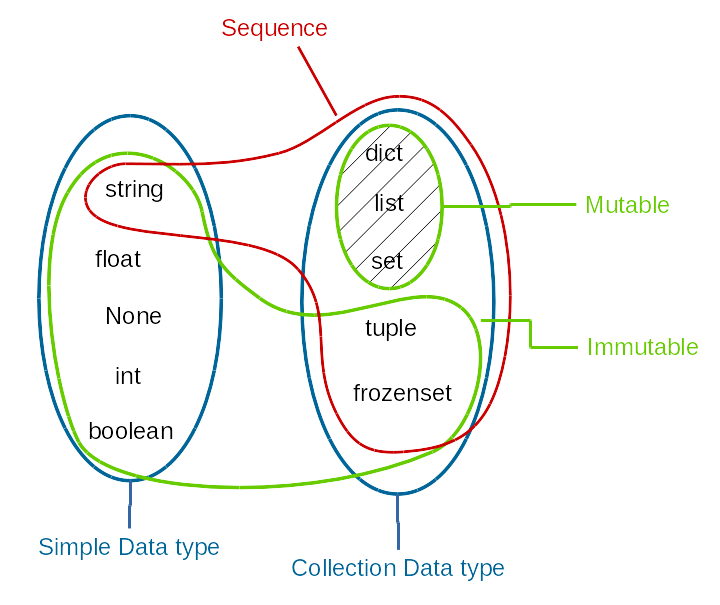
Sequences¶
Sequence types share common operations. We have already encoutered the string type. All the operations below can be used on strings and other sequence types, which we will cover soon.
| Operation | Result |
|---|---|
x in s |
True if an item of s is
equal to x, else False |
x not in s |
False if an item of s is
equal to x, else True |
s + t |
the concatenation of s and t |
s * n ( or
n * s) |
equivalent to adding s to itself n times |
s[i] |
ith item of s, origin 0 |
s[i:j] |
slice of s from i to j |
s[i:j:k] |
slice of s from i to j with step k |
len(s) |
length of s |
min(s) |
smallest item of s |
max(s) |
largest item of s |
s.index(x[, i[, j]]) |
index of the first occurrence of x in s (at or after index i and before index j) |
s.count(x) |
total number of occurrences of x in s |
Example1: get the length of a sequence
s = "Protein"
len(s)
Example2: add (concatenate) two strings
new_string = "abc" + "def"
new_string
Accessing items and slicing sequences¶
- Accessing to an item uses the square brackets syntax
- Index starts at 0
- Index ends at N-1 , where N is the length of the sequence
- [index] get the item at position defined by index
s = "Protein"
print(s[0])
print(s[3])
- Slicing can access to a subset of the sequence (not just one item)
- Slicing uses the same square brackets syntax as above and should contain a : character. See syntax here below
- [start:end:step] most general slicing from start to end (excluded) with a step - [start:end] (step=1) - [start:] (step=1, until the sequence end) - [:] (start=0, until the sequence end, step=1) - [::2] (start=0, until the sequence end, step=2)
- [start:end:step] most general slicing from start to end - [start:end] (step=1) - [start:] (step=1, until the sequence end) - [:] (start=0, until the sequence end, step=1) - [::2] (start=0, until the sequence end, step=2)
# Then [start:end] syntax
s[1:4]
# If the end is not provided, its default is -1 (up to the end)
s[2:]
# it the start is omitted, it defaults to 0
s[:2]
# We can omit start and end
s[:]
# Or select every two elements
s[::2]
In Python, indices can also be negatives. Consider this example on the string Protein.
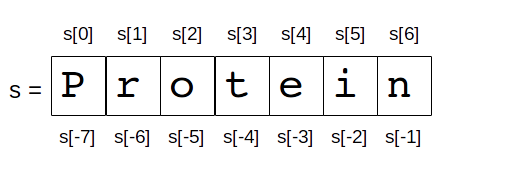
# slice with negative
s[-3:-1]
s[1:-1:2]
s[::2]
s[::-2]
s[::-1]
Strings are immutable sequences¶
s = "Protein"
s[3] = 'Z'
# to change an item, you got to create a new string
s2 = s[:-1] + 'c'
print(s2)
String methods¶
print(dir("")[32:])
Well, that's a lot ! Here are some useful ones:
| Method | Example |
|---|---|
| find | s1.find("GGG") |
| index | s1.index("GGG") |
| replace | s1.replace("T", "U") |
| lower | s1.lower() |
| split | s1.split("\n") |
| endswith | s1.endswith("n") |
| startswith | s1.startswith("n") |
| strip | s1.strip() |
The split and join method (on string sequences)¶
There are many methods that can be used with strings. A pair of methods s worth mentionning here: the split and join.
Consider the following sentence. You want to extract each word. The unique separator is a space.
sentence = "This is amazing " # note the extra space
# By default, the split method splits the string on whitespace
words = sentence.split()
words
The returned object is a sequence of strings. It is a new type of sequence: the list, which will be covered later.
You can now join back the 3 strings. You can join them with a space to recover the orignal sentence, or another character (e.g. return carriage, denoted \n) or a longer string:
" -- ".join(words)
The in and not keywords¶
sequence = "ACGTAAAA"
"N" in sequence
"A" in sequence
"A" not in sequence
not True
String manipulation
data = open("data/P56945.fasta").read()
- The data variable is a string. It contains a header and a protein sequence. Print the data. Not the return carriage. Extract the header (first line) and the entire sequence without the \n signs
- Get the length of the sequences.
- Get position (index) of the accession number (P56945) from the header
- Extract the accession number
- Extract the uniprot identifier (BCAR1_HUMAN)
- What are the starting position of the following sequences: PPAEREAPAE and PPPAAA
sv40 = open("data/sv40.fasta").read()
- The sv40 variable is a string. genome sequence of sv40.
- Get the length of the sequences.
- Can you find following recognition sites in the sequence ?:
- BamHI (ggatcc),
- EcorI (gaattc),
- HindIII (aagctt),
- SmaI (cccggg) (answer by True or False)?
- for the enzymes which have a recognition site can you give their positions? is there only one site in sv40 per enzyme?
- compute the GC percent of sv 40
We want to perform a PCR on sv40, can you give the length and the sequence of the amplicon?
primer_1 : 5' CGGGACTATGGTTGCTGACT 3'
primer_2 : 5' TCTTTCCGCCTCAGAAGGTA 3'
The tuple sequence¶
A tuple is an ordered and immutable sequence of arbitrary items
To create a tuple, use:
- the tuple( ) builtin function
- a comma after an item
- parentheses with at least 1 comma
Examples¶
(1, 2, 3)
tuple([1,'a',3])
(1) # Note: braces does not create a tuple, only the comma does.
(1,)
Immutable¶
t = (1,2,3)
t[0] = 1
Some operators¶
(1,) * 5
# You can actually append item with the augmented operator
# but += creates a new object
t1 = (1, 0)
t1 += (1, 2)
t1
t1 *= 2
t1
Tuple methods¶
dir(tuple())[iN:]
Only two methods !! Much simpler than strings because tuples are optimised for speed
t = (1, 2, 1, 2, 2, 2, 7, 5)
t.count(2)
t.index(5)
Tuples can be use to swap elements¶
a = 2
b = 10
a, b, = b, a
a, b
The list sequence¶
A list is an ordered and mutable sequence of arbitrary items
To create a list, use:
- the list( ) builtin function
- square brackets [ ]
Examples¶
l2 = [1, 'a', 3] # # you can have any kind of mixed objects in a lists.
print(l2)
# slicing and indexing like for strings are available
l2[1]
l2[0::2]
list("sequence")
Lists are Mutable sequences¶
l2 = [1, 'a', 3]
l2[1] = 2
l2
Some handy operators¶
[1, 2] + [3, 4]
[1] * 10
ll = [1,2,3]
ll += [4,5,6]
ll
List methods¶
print(dir(methods)[iN:])
Difference between append and extend¶
Lists have several methods amongst which the append and extend methods. The former appends an object to the end of the list (e.g., another list) while the later appends each element of the iterable object (e.g., another list) to the end of the list.
stack = ['a','b']
stack.append('foo')
stack
However, if we want to append several objects contained in a list, the result as expected (or not...) is:
stack.append(['d', 'e', 'f'])
stack
To include each element, you should use the extend method instead of append
stack = ['a', 'b', 'c']
stack.extend(['d', 'e','f'])
stack
stack = ['a','b']
stack.extend('foo')
stack
stack = ['a', 'b', 'c']
stack.extend(['d', 'e', ['f']])
stack
Tuples VS lists¶
- Tuples are faster than list (see examples at the end of this notebook)
- Tuples protect the data (immutable)
- Tuples can be used as keys on dictionaries, not lists.
- Tuples can be added to a set (see later), not lists.
List practical session
- Create a list of 40 letters made of 10 A, 10 C, 10 G, 10 T
- Select elements between index 10 (included) and 20 (included). What is the length of the new list. Check you only have C letters.
- Select only the G letters using slicing
- count the number of T
- Create two lists [1,2,3,4,5] and [6,7,8,9]
- concatenate them using the + operator. concatenate them using the extend method. What the fundamental difference ?
From the list l = [1, 2, 3, 4, 5, 6, 7, 8, 9] generate 2 lists l1 containing all odd values, and l2 all even values.
l = [1, 2, 3, 4, 5, 6, 7, 8, 9] l1 = lxxx l2 = lxxx print(l1) [1,3,5,7,9] print(l2) [2,4,6,8]
Given
l = [1, 2, "a"] t = (1,2)check that a is included in the list. What about the tuple t, is it included in l ?
The dictionary sequence (dict)¶
a dictionary is an unordered and mutable collections of key–value items
- It is equivalent to a mapping type where we associate an item to a key
- Dictionary are fast because a key provide a direct access to its corresponding item (the value), without the need to iterate through the entire collection
Examples¶
d = {} # empty dictionary
d = {'first':1, 'second':2} # initialise a dictionary
# access to value given a key:
d['first']
# add a new pair of key/value:
d['third'] = 3
# what are the keys ?
d.keys()
# what are the values ?
d.values()
Mutable¶
d['third'] = [3,3,3,3]
d
Dictionary methods¶
d = {}
methods = dir(d)
print(methods[28:])
Dictionary Practical session
data = open("data/P56945.fasta").read()
- Extract the entire sequence as a single string (already done, but do it again)
- Count the number of A, C, G, T
- Create a dictionary d1 with four keys: A, C, G, T and their corresponding number of occurences
- Create a new dictionary d2 with one item whose key is "N" and value is 0
- Create a new dictionary that combines d1 and d2
- Starting from
Change the content of d4 (e.g., change the value corresponding to the key a). Check the content of d3. Did it change ?d3 = {"a":1, "b":2} d4 = d3- Using d3 from above, try to access to a key that does not exist (e.g., c). What is happening ? How to check that c does not exit so as to avoid the error ?
- Try to create a dictionary with a list [1,2] as a key. Do the same with a tuple.
set sequences¶
A set is an unordered and mutable sequence of unique items
- Sets are mutable, so we can easily
addorremoveitems. - items are unique
- Since sets are unordered they have no notion of index position (no slicing)
The syntax to create a set can be the function set or curly braces {}
a = {1, 2, 3, 4, 4}
a
a = set([1, 2, 3, 4, 4]) # input is a list
a
b = set((3, 4, 5, 6)) # input is a tuple
b
sets are mutable¶
s = set()
print(s)
s.add('a')
print(s)
s.add('b')
print(s)
s.add((1,2))
print(s)
set also support set operators: Union, Intersection, Difference, Symetric difference
| operator | description |
|---|---|
| | | for union |
| & | for intersection |
| < | for subset |
| - | for difference |
| ^ | for symmetric difference |
a | b
a & b
a < b
a - b
a ^ b
These operators are available as methods as well. E.g,
a.difference(b)
a.union(b)
frozensets (for information)¶
Sets are mutable, and may therefore not be used, for example, as keys in dictionaries.
Another problem is that sets themselves may only contain immutable (hashable) values, and thus may not contain other sets.
Because sets of sets often occur in practice, there is the frozenset type, which represents immutable (and, therefore, hashable) sets.
a = frozenset({1,2,3})
a
a = set([1,2,3])
b = set([2,3,4])
# a.add(b) # this is NOT possible since b is mutable
a.add(frozenset(b))
a
A note about copying collections¶
- everything is an object and therefore references are used !!
- consider the following example
a = [1, 2, 3]
b = a # is a reference
b[0] = 10
print(a)
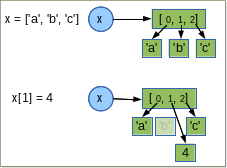
In some situations, we really do want a separate copy of the item, or collections of objects.
With sequences, you can use slicing. Indeed, the slice is always an independent copy of the items copied.
To copy an entire sequence we can do this:
ascii = ['a','b','c']
ascii_copy = ascii[:]
For dictionaries and sets, copying can be achieved using dict.copy( ) and set.copy( ).
shallow versus deep copy¶
Using slicing to copy the list: no surpise
ascii = ['a','b','c']
ascii_copy = ascii[:] # shallow copy
ascii[2] = 'z'
print("ascii", ascii)
print("ascii_copy", ascii_copy)
If an item is a sequence itself (here ['c', 'd']), it is copied but the items inside are independent objects, so they are not copied (reference).
ascii = ['a','b',['c', 'd']]
ascii_copy = ascii[:]
ascii_copy[2][0] = "eeee"
print("ascii", ascii)
print("ascii_copy", ascii_copy)
Solution: use copy module and the deepcopy function
import copy
ascii = ['a', 'b', ['c', 'd']]
ascii_copy = copy.deepcopy(ascii)
ascii_copy[2][0] = "eeee"
ascii
Writing into a file¶
mysequence = """>sp|P56945|BCAR1_HUMAN Breast cancer anti-estrogen resistance protein 1 OS=Homo sapiens GN=BCAR1 PE=1 SV=2
MNHLNVLAKALYDNVAESPDELSFRKGDIMTVLEQDTQGLDGWWLCSLHGRQGIVPGNRL
KILVGMYDKKPAGPGPGPPATPAQPQPGLHAPAPPASQYTPMLPNTYQPQPDSVYLVPTP
SKAQQGLYQVPGPSPQFQSPPAKQTSTFSKQTPHHPFPSPATDLYQVPPGPGGPAQDIYQ
VPPSAGMGHDIYQVPPSMDTRSWEGTKPPAKVVVPTRVGQGYVYEAAQPEQDEYDIPRHL
LAPGPQDIYDVPPVRGLLPSQYGQEVYDTPPMAVKGPNGRDPLLEVYDVPPSVEKGLPPS
NHHAVYDVPPSVSKDVPDGPLLREETYDVPPAFAKAKPFDPARTPLVLAAPPPDSPPAED
VYDVPPPAPDLYDVPPGLRRPGPGTLYDVPRERVLPPEVADGGVVDSGVYAVPPPAEREA
PAEGKRLSASSTGSTRSSQSASSLEVAGPGREPLELEVAVEALARLQQGVSATVAHLLDL
AGSAGATGSWRSPSEPQEPLVQDLQAAVAAVQSAVHELLEFARSAVGNAAHTSDRALHAK
LSRQLQKMEDVHQTLVAHGQALDAGRGGSGATLEDLDRLVACSRAVPEDAKQLASFLHGN
ASLLFRRTKATAPGPEGGGTLHPNPTDKTSSIQSRPLPSPPKFTSQDSPDGQYENSEGGW
MEDYDYVHLQGKEEFEKTQKELLEKGSITRQGKSQLELQQLKQFERLEQEVSRPIDHDLA
NWTPAQPLAPGRTGGLGPSDRQLLLFYLEQCEANLTTLTNAVDAFFTAVATNQPPKIFVA
HSKFVILSAHKLVFIGDTLSRQAKAADVRSQVTHYSNLLCDLLRGIVATTKAAALQYPSP
SAAQDMVERVKELGHSTQQFRRVLGQLAAA
"""
# First, we open a file in "w" write mode
fh = open("mysequence.fasta", "w")
# Second, we write the data into the file:
fh.write(mysequence)
# Third, we close:
fh.close()
Reading data from a file¶
# First, we open the file in read mode (r)
fh = open('mysequence.fasta', 'r')
# Second, we read
data = fh.read()
# Third we close
fh.close()
# data is now a string that contains the content of the file being read
print(data)
File manipulation
- Read data/P56945.fasta file and save the content in a variable.
- Save into another file. Read back the new file and get the sequence; save into seq2
- check that seq1 and seq2 are identical
- count the number of A (88)
- count the number of C, G, T and save into a dictionary where letters are keys and counts are the values
For advanced users: get at least two different uniprot identifiers and download the sequences from uniprot website (see first slides).
Get the ACGT letter counts for the different sequences.
Save results as dictionary of dictionaries so that we can get the number of A of uniprot ID1 as results['ID1']['A']
The with keyword¶
- When dealing with files, it is important to close the file handler.
- A special keyword with can be used and you will see the following syntax quite often
- Once used with the with keyword, the file handler will be automatically closed.
- Here is the syntax
with open("mysequence.fasta", "r") as fh:
data = fh.read()
data
There is some magic here since fh has been closed automatically. Ther with keywords is a Context Manager.
The del keyword¶
my_variable = 1
my_variable
del(my_variable)
my_variable
Summary¶
- Python language uses indentation to create block of statements. The convention is to use 4 spaces (not tabulation)
- We've seen some Scalar types: float, complex, long
- Scalar types can be created on the fly or with built-in functions:
var_int = 1
var_int = int(1.1)- Variable names can be made of characters and underscores but avoid starting with underscores except if you know what you are doing
Python has reserved keywords that cannot be used as variable name. E.g:
from, import, as, print, del, with, in, not
- dot operator: allows one to access to content of any object
- Python has a few built-in functions.
- print is one of the most used built-in function
- int( ) , float( ), tuple( ), list( ), dir( ), set( ) , dict( ), open( ), close( ) ...
- We've seen how to import a module using the following keywords: import/from/as
- del removes an object from the environment.
We've seen the most important data structures in Python:
lists are mutable sequences:
l = [1,2,3]
tuples are immutable sequences:
t = (1,2,3)
dictionaries are hash tables :
d = {'a': 1, 'b': 2}
sets are unordered sequences with unique values
s = set([1,2,3])
- Be aware that mutable objects (e.g., list) are copied by reference:
a = [1,2,3]
b = a
When changing b you are also changing a
If you really want a different object, copy it for instance by slicing its contents:
b = a[:]- Use tuple instead of list whenever possible (more efficient)
- When you want to use membership operator in, use set instead of list whenever possible (more efficient).