Object-oriented Architecture and Design¶
Inheritance¶
In the introduction of I mentioned that one of the objectives of OOP is to address some of the issues of software quality.
What we have seen so far, object-based programming, consists in designing programs with objects, that are built with classes. In most object-oriented programming languages, you also have a mechanism that enables classes to share code. The idea is very simple: whenever there are some commonalities between classes, why having them repeat the same code, thus leading to maintenance complexity and potential inconsistency? So the principle of this mechanism is to define some classes as being the same as other ones, with some differences.
In the example below we have design two classes to represent to kind of sequences DNA or AA. As we can see the 2 kinds of sequence have the same attributes (name and sequence) have some common methods (len, to_fasta) but have also some specific methods gc_percent and molecular_weight.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 | from __future__ import print_function
class DNASequence(object):
alphabet = 'ATGC'
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def to_fasta(self):
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
def gc_percent(self):
return float(self.sequence.count('G') + self.sequence.count('C')) / len(self.sequence)
class AASequence(object):
_water = 18.0153
_weight_table = {'A': 89.0932, 'C': 121.1582, 'E': 147.1293,
'D': 133.1027, 'G': 75.0666, 'F': 165.1891,
'I': 131.1729, 'H': 155.1546, 'K': 146.1876,
'M': 149.2113, 'L': 131.1729, 'O': 255.3134,
'N': 132.1179, 'Q': 146.1445, 'P': 115.1305,
'S': 105.0926, 'R': 174.201, 'U': 168.0532,
'T': 119.1192, 'W': 204.2252, 'V': 117.1463,
'Y': 181.1885}
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def to_fasta(self):
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
def molecular_weight(self):
return sum([self._weight_table[aa] for aa in self.sequence]) - (len(self.sequence) - 1) * self._water
|
The problem with this implementation is that a large part of code is the same in the 2 classes. It’s bad because if I have to modify a part of common code I have to do it twice. If in future I’ll need a new type of Sequence as RNA sequence I will have to duplicate code again on so on. the code will be hard to maintain. I need to keep together the common code, and be able to specify only what is specific for each type of Sequences
So we keep our two classes to deal with DNA and protein sequences, and we add a new class: Sequence, which will be a common class to deal with general sequence functions. In order to specify that a DNA (or a Protein) is a Sequence in Python is:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 | from __future__ import print_function
class Sequence(object):
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def to_fasta(self):
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
class DNASequence(Sequence):
alphabet = 'ATGC'
def gc_percent(self):
return float(self.sequence.count('G') + self.sequence.count('C')) / len(self.sequence)
class AASequence(Sequence):
_water = 18.0153
_weight_table = {'A': 89.0932, 'C': 121.1582, 'E': 147.1293,
'D': 133.1027, 'G': 75.0666, 'F': 165.1891,
'I': 131.1729, 'H': 155.1546, 'K': 146.1876,
'M': 149.2113, 'L': 131.1729, 'O': 255.3134,
'N': 132.1179, 'Q': 146.1445, 'P': 115.1305,
'S': 105.0926, 'R': 174.201, 'U': 168.0532,
'T': 119.1192, 'W': 204.2252, 'V': 117.1463,
'Y': 181.1885}
def molecular_weight(self):
return sum([self._weight_table[aa] for aa in self.sequence]) - (len(self.sequence) - 1) * self._water
|
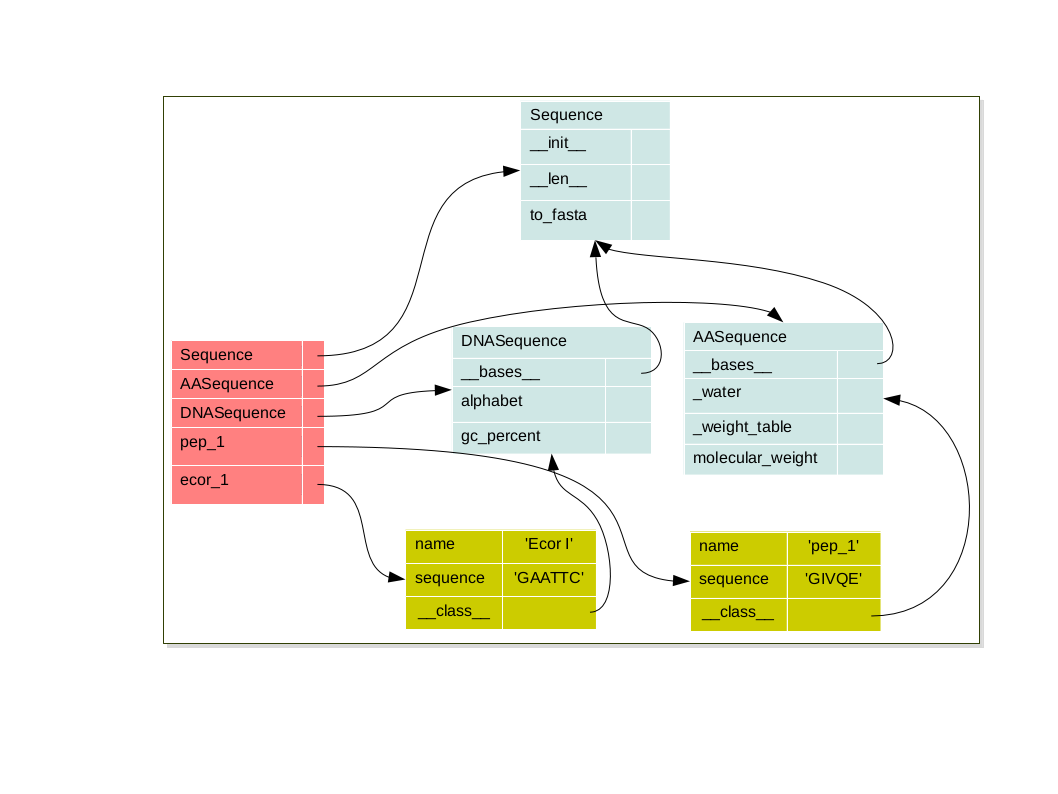
- Each object have it’s own namespace which are linked to the class namespace via the special attribute:
__class__ - Each class is link to it’s parents namespace via the special attribute
__bases__on so on until the object class namespace
pep_1 = AASequence('pep_1', 'GIVQE')
bar = DNASequence('Ecor I', 'GAATTC')

Overloading¶
Overloading an attribute or a method is to redefine at a subclass level an attribute or method that exists in upper classes of a class hierarchy.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 | from __future__ import print_function
class Sequence(object):
_water = 18.0153
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def to_fasta(self):
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
def molecular_weight(self):
return sum([self._weight_table[x] for x in self.sequence]) - (len(self.sequence) - 1) * self._water
class DNASequence(Sequence):
alphabet = 'ATGC'
_weight_table = {'A': 331.2218, 'C': 307.1971, 'G': 347.2212, 'T': 322.2085}
def gc_percent(self):
return float(self.sequence.count('G') + self.sequence.count('C')) / len(self.sequence)
def _one_strand_molec_weight(self, seq):
return sum([self._weight_table[base] for base in seq]) - (len(seq) - 1) * self._water
def molecular_weight(self):
direct_weight = self._one_strand_molec_weight(self.sequence)
rev_comp = self.rev_comp()
rev_comp_weight = self._one_strand_molec_weight(rev_comp.sequence)
return direct_weight + rev_comp_weight
class RNASequence(Sequence)
alphabet = 'AUGC'
_weight_table = {'A': 347.2212, 'C': 323.1965, 'G': 363.2206, 'U': 324.1813}
class AASequence(Sequence):
_weight_table = {'A': 89.0932, 'C': 121.1582, 'E': 147.1293,
'D': 133.1027, 'G': 75.0666, 'F': 165.1891,
'I': 131.1729, 'H': 155.1546, 'K': 146.1876,
'M': 149.2113, 'L': 131.1729, 'O': 255.3134,
'N': 132.1179, 'Q': 146.1445, 'P': 115.1305,
'S': 105.0926, 'R': 174.201, 'U': 168.0532,
'T': 119.1192, 'W': 204.2252, 'V': 117.1463,
'Y': 181.1885}
|
We we overload a method sometimes we just want to add something to the parent’s method. in this case we can call
explicitly the parent’s method by using the keywords super. The syntax of this method is lit bit tricky.
the first argument must be the class that you want to retrieve the parent (usually the class you are coding),
the second argument is the object you want to retrieve the parent class (usual self) and it return a proxy to the parent
so you just have to call the method. see it in action, in the example below we overload the __init__ method
and just add 2 attribute but for the name and sequence we call the Sequence __init__ method.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 | from __future__ import print_function
class Sequence(object):
_water = 18.0153
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def to_fasta(self):
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
def molecular_weight(self):
return sum([self._weight_table[x] for x in self.sequence]) - (len(self.sequence) - 1) * self._water
class RestrictionEnzyme(Sequence):
def __init__(self, name, seq, cut, end):
# the line below is in python3 only
super().__init__(name, seq)
# in python2 the syntax is
# super(RestrictionEnzyme, self).__init__(name, seq)
# this syntax is also available in python3
self.cut = cut
self.end = end
ecor1 = DNASequence('ecor I', 'GAATTC', 1, "sticky")
print(ecor1.name, len(ecor1))
|
In python3 the syntax has been simplified. we can just call super() that’s all.
Polymorphism¶
The term polymorphism, in the OOP lingo, refers to the ability of an object to adapt the code to the type of the data it is processing.
Polymorphism has two major applications in an OOP language. The first is that an object may provide different implementations of one of its methods depending on the type of the input parameters. The second is that code written for a given type of data may be used on other data with another datatype as long as the other data have compatible behavior.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 | from __future__ import print_function
def my_sum(a, b):
return a + b
print("my_sum(3, 4) =", my_sum(3, 4))
print("my_sum('three', 'four') =", my_sum('three', 'four'))
class Sequence(object):
_water = 18.0153
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
return len(self.sequence)
def __str__(self):
return ">{}\n{}".format(self.name, self.sequence)
def __add__(self, other):
return Sequence('{}/{}'.format(self.name, other.name),
self.sequence + other.sequence)
ecor_1 = Sequence('Ecor_I', 'GAATTC')
bamh_1 = Sequence('Bamh I', 'GGATCC')
print("my_sum(ecor_1, bamh_1) =", my_sum(ecor_1, bamh_1))
|
Albeit data are type, my method my_sum work equally on different type as integer, string or sequence. The my_sum method is called polymorph.
Multiple inheritance¶
In Python, as in several programming language, you can have a class inherit from several base classes. Normally, this happens when you need to mix very different functionalities.
Inheriting from classes that come from the same hierarchy can be tricky, in particular if methods are defined everywhere: you will have to know how the interpretor choose a path in the classes graph. But this case is more like a theoretical limit case, that should not happen in a well designed program.
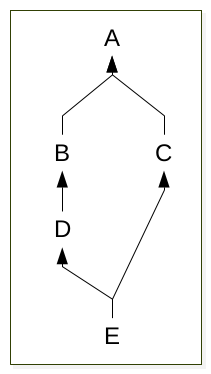
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | from __future__ import print_function
class A(object):
def met_1(self):
print("A.meth_1")
class B(A):
pass
class C(B):
pass
class D(B):
def met_1(self):
print("D.meth_1")
class E(C, D):
pass
e = E()
e.met_1()
|
Which method will be executed?
To determine the order of lookup, python use an algorithm call method resolution order .
in the example above, the result of mro is:
>>> E.__mro__
(<class '__main__.E'>, <class '__main__.C'>, <class '__main__.D'>, <class '__main__.B'>, <class '__main__.A'>, <class 'object'>)
Abstract classes¶
An abstract class is a class which contains some abstract methods. A such class cannot be instantiated. A class that has a metaclass derived from ABCMeta cannot be instantiated unless all of its abstract methods and properties are overridden.
By defining an abstract base class, you can define a common API for a set of subclasses. This capability is especially useful in situations where a third-party is going to provide implementations, such as with plugins to an application, but can also aid you when working on a large team or with a large code-base where keeping all classes in your head at the same time is difficult or not possible.
Note
Unlike Java abstract methods, these abstract methods may have an implementation. This implementation can be called via the super() mechanism from the class that overrides it. This could be useful as an end-point for a super-call in a framework that uses cooperative multiple-inheritance.
example of abstract class:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 | from abc import ABCMeta, abstractmethod
# the syntax below is python3 specific
# for python 2 use the following syntax
#
# class Sequence(object)
#
# __metaclass__ = ABCMeta
#
class Sequence(object, metaclass=ABCMeta):
_water = 18.0153
def __init__(self, name, seq):
"""
:param seq: the sequence
:type seq: string
"""
self.name = name
self.sequence = seq
def __len__(self):
"""
:return: the length of this sequence (number of bases or aa)
:rtype: integer
"""
return len(self.sequence)
def to_fasta(self):
"""
:return: a string in fasta format of this sequence
:rtype: basestring
"""
id_ = self.name.replace(' ', '_')
fasta = '>{}\n'.format(id_)
start = 0
while start < len(self.sequence):
end = start + 80
fasta += self.sequence[start: end + 1] + '\n'
start = end
return fasta
def _one_strand_molec_weight(self, seq):
"""
helper function to compute the mw of the seq
:param seq: the seq (nucleic or proteic) to compute the mw
:type seq: string
:return: mw of seq
:rtype: float
"""
return sum([self._weight_table[base] for base in seq]) - (len(seq) - 1) * self._water
@abstractmethod
def molecular_weight(self):
"""
tihis method is abstract and must be implemented in child classes
:return: The molecular weight
:rtype: float
"""
pass
@property
def alphabet(self):
return self._alphabet
class DNASequence(Sequence):
_alphabet = 'ACGT'
_weight_table = {'A': 347.2212, 'C': 323.1965, 'G': 363.2206, 'T': 322.2085}
def gc_percent(self):
"""
:return: the ratio of G and C bases in sequence
:rtype: float
"""
return float(self.sequence.count('G') + self.sequence.count('C')) / len(self.sequence)
def molecular_weight(self):
"""
:return: The molecular weight
:rtype: float
"""
direct_weight = super()._one_strand_molec_weight(self.sequence)
rev_comp = self.rev_comp()
rev_comp_weight = super()._one_strand_molec_weight(rev_comp.sequence)
return direct_weight + rev_comp_weight
def rev_comp(self):
"""
:return: a new sequence representing the reverse complement
:rtype: :class:`DNASequence` object
"""
rev = self.sequence[::-1]
table = str.maketrans(self._alphabet, self._alphabet[::-1])
rev_comp = str.translate(rev, table)
return DNASequence(self.name + '_reverse', rev_comp)
class AASequence(Sequence):
_water = 18.0153
_weight_table = {'A': 89.0932, 'C': 121.1582, 'E': 147.1293,
'D': 133.1027, 'G': 75.0666, 'F': 165.1891,
'I': 131.1729, 'H': 155.1546, 'K': 146.1876,
'M': 149.2113, 'L': 131.1729, 'O': 255.3134,
'N': 132.1179, 'Q': 146.1445, 'P': 115.1305,
'S': 105.0926, 'R': 174.201, 'U': 168.0532,
'T': 119.1192, 'W': 204.2252, 'V': 117.1463,
'Y': 181.1885}
_alphabet = ''.join(_weight_table.keys())
def molecular_weight(self):
"""
:return: The molecular weight
:rtype: float
"""
return super()._one_strand_molec_weight(self.sequence)
if __name__ == '__main__':
nuc = DNASequence('Ecor I', 'GAATTC')
prot = AASequence('my prot', 'MLEFVQ')
for molec in [nuc, prot]:
print(molec.name, molec.molecular_weight())
print(molec.name, molec.alphabet)
print('-' * 10)
neither_nuc_nor_prot = Sequence('truc', 'GAATTC')
|
python3 abstract_sequence.py
Ecor I 3870.4
Ecor I ACGT
----------
my prot 765.9168999999999
my prot VFHNWARLQCSDTOEMUPYKGI
----------
Traceback (most recent call last):
File "abstract_sequence.py", line 132, in <module>
neither_nuc_nor_prot = Sequence('truc', 'GAATTC')
TypeError: Can't instantiate abstract class Sequence with abstract methods molecular_weight
Composition¶
Here is a problem that come to me during a software design. I have to modelize a generic macromolecular system of proteins. following the main characteristics of a system.
- A systems is composed of several genes
- some genes a mandatory
- some genes are accessory
- some other are forbidden
- for some efficient reason each genes must search only ones
I have in some case to search for lot of systems in on run.
Ok I have only each gene is unique for all systems. So I design the program to have only one instance of a gene. but several systems can reference the same gene. after some experiments it was obvious we need a new concept to be more sensitive.
- some genes have homologs and some other have analogs
- a gene can appeared in one system a mandatory gene and is an homolog of an other gene in an other system.
- a homologs have the same characteristics than a gene, it just have in more the reference to the gene it is the homologs
- the analogs are the same as homologs.
The first idea which came in my mind was to create an Homolog and Analog classes which inherits of Gene. But if I did that when a gene appeared as gene in a system and as homologs in other systems. I search this gene twice. It was not I want. So I need to think again to my design. I decide to use composition instead of inheritance. I create a class of Analog and Homologue but which not inherits from Gene. But they have a property gene encapsulated inside them. And I use delegation for all properties and methods for gene. This means that I create same properties in Homologs that exist in Gene, but when I call these property in homolgs it just call the method from the gene encapsulated.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 |
Class Gene:
def __init__(self, name, system, loner, profile):
self.name = name
self.profile = profile
self._system = system
self._loner = loner
@property
def system(self):
"""
:return: the System that owns this Gene
:rtype: :class:`macsypy.system.System` object
"""
return self._system
def __eq__(self, gene):
"""
:return: True if the gene names (gene.name) are the same, False otherwise.
:param gene: the query of the test
:type gene: :class:`macsypy.gene.Gene` object.
:rtype: boolean.
"""
return self.name == gene.name
class Homolog:
def __init__(self, name, system, loner, profile, gene_ref, aligned=False):
super(Homolog, self).__init__(name, system, loner, profile)
self.ref = gene_ref
self.aligned = aligned
@property
def system(self):
"""
:return: the System that owns this Gene
:rtype: :class:`macsypy.system.System` object
"""
return self.gene.system
def __eq__(self, gene):
"""
:return: True if the gene names (gene.name) are the same, False otherwise.
:param gene: the query of the test
:type gene: :class:`macsypy.gene.Gene` object.
:rtype: boolean.
"""
return self.gene.name == gene.name
def is_aligned(self):
"""
:return: True if this gene homolog is aligned to its homolog, False otherwise.
:rtype: boolean
"""
return self.aligned
|
This design pattern is called composition. This is a powerful design that mimics inheritance but with more flexibility.
But it’s lit bit boring to recode all methods we need to delegate to the gene. So in python there is a special
method which allow to do this automatically: __getattr__ when python look inside the instance and it does not find
an attribute (remember in python methods are attributes) and find the special method __getattr__ it call this method
with the looking attribute as argument.

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 |
Class Gene:
def __init__(self, name, system, loner, profile):
self.name = name
self.profile = profile
self._system = system
self._loner = loner
@property
def system(self):
"""
:return: the System that owns this Gene
:rtype: :class:`macsypy.system.System` object
"""
return self._system
def __eq__(self, gene):
"""
:return: True if the gene names (gene.name) are the same, False otherwise.
:param gene: the query of the test
:type gene: :class:`macsypy.gene.Gene` object.
:rtype: boolean.
"""
return self.name == gene.name
class Homolog:
def __init__(self, gene, gene_ref, aligned=False):
self.gene = gene
self.ref = gene_ref
self.aligned = aligned
def __getattr__(self, name):
return getattr(self.gene, name)
def is_aligned(self):
"""
:return: True if this gene homolog is aligned to its homolog, False otherwise.
:rtype: boolean
"""
return self.aligned
|
Warning
It is very convenient but you must use __getattr__ with parsimony and well document it as it can obfuscate the code.
Exercises¶
Exercise¶
Create 2 classes
- Sequence
- MutableSequence
These 2 classes have same attributes but Sequence can be mutate and extend whereas Sequence are immutable.
Exercise¶
how can you modeling
- non mutable DNA sequence
- mutable DNA sequence
- non mutable amino acid sequence
- mutable amino acid sequence
can you easily extend your model to support no mutable/ mutable RNA sequence ?
Note
You have not to implement all methods just design the api: the class, attributes, and methods
Exercise¶
work with in small groups.
We want to implement a annotation de novo assembled contigs pipeline. We used BLAST to compare our contigs to sequence databases in order to annotate them with similarity to known genes/proteins/functions. By querying three major databases,
- GenBank’s non-redundant protein database (NR)
- Uniprot’s Swiss-Prot
- TrEMBL protein databases,
We will identify the most similar known sequences for each of our contigs. The available information about these matching sequences will be used to annotate our contigs with likely functional properties. For the significant matches in the database, we will extract both gene names, general descriptions, and Gene Ontology (GO) categories (specific categorical classifications grouping genes based on cellular and molecular function, e.g. “cellular response to protein unfolding” or “calcium homeostasis”), along with additional information from the Uniprot knowledge database.
Do not implement methods. Just draw a class diagram of this application.
Note
Class Diagram provides an overview of the target system by describing the objects and classes inside the system and the relationships between them.
example of class diagram in uml formalism: