Searching In Files
grep
print lines matching a pattern
- -i (--ignore-case) Ignore case distinctions in both the PATTERN and the input files.
- -v (--invert-match) Invert the sense of matching, to select non-matching lines.
- -r (--recursive) Read all files under each directory, recursively, following symbolic links only if they are on the command line.
- -n (--line-number) Prefix each line of output with the 1-based line number within its input file.
FASTA file format
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
FASTQ file format
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Exercises
- count the number of sequences in the file 'abcd.fa' (a multiple fasta file)
- can we have the same approach with a fastq file?
grep with Regular Expressions
- grep -E or egrep
- Regular Expressions: sequences of characters to express a pattern
- useful metacharacters:
- . any character
- [] bracket expressions
- ^ start of the string
- $ end of the string
- * match 0 to n times the preceding element
- {m,n} match m to n times the preceding element
- (one|two) match the strings "one" or "two"
Exercises
- find the names of all the genes in the zipped FASTA file abcd.fa.gz in the unix_training project directory of central-bio.
- in the file abcd_sequences.gb, in the unix_training project directory of central-bio, find the origin organism of all the sequence entries (tip: it's on a line with the ORGANISM keyword).
Filtering/sorting results
cut
- cut - remove sections from each line of files
cut OPTION... [FILE]- -d DELIM use DELIM instead of TAB for field delimiter
- -f select only these fields
who | cut -d ' ' -f 1
find . -atime 0 | cut -d '/' -f 2,3
sort
- sort - sort lines of text files
sort [OPTION]... [FILE]- -k --key=KEYDEF sort via a key, KEYDEF gives location and type.
- -n (--numeric-sort) compare according to string numerical value (OBSOLETE).
- -r (--reverse) reverse the result of comparisons.
who | cut -d ' ' -f 1 | sort | uniq -c | sort -k 1n,2
Exercise
We ran a blast with -m8 output. So the following fields are displayed
id, percent identity, alignment length, number of mismatches,
number of gap openings, query start, query end,
subject start, subject end,
Expect value, HSP bit score
Exercise (continued)
separated by tab.
- copy the file blast2_m8.txt in the project of the course in your home.
- sort the output folowing the % of identity (the highest identity to the top)
- display only columns id of hit, percent identity, Expect value, HSP bit score
- store the results in a new file.
uniq
report or omit repeated lines (Filter adjacent matching lines)
- uniq [OPTION] INPUT
- -c prefix lines by the number of occurrences.
Exercise
- eliminate duplicates from the list of organisms gathered from abcd_sequences.gb.
- from the same blast output than previous exercise, display the sequence ids of the match.
xargs
build and execute command lines from standard input xargs executes the command with any initial-arguments followed by items read from standard input.
Do not forget to add -l to process standard input line by line
find . -name '*.fasta' | xargs grep ">"
- -I allows you to insert the line from the standard input somewhere else than in the end of the command
find . -name '*.fasta' | xargs -I fic cat fic >>allmysequences
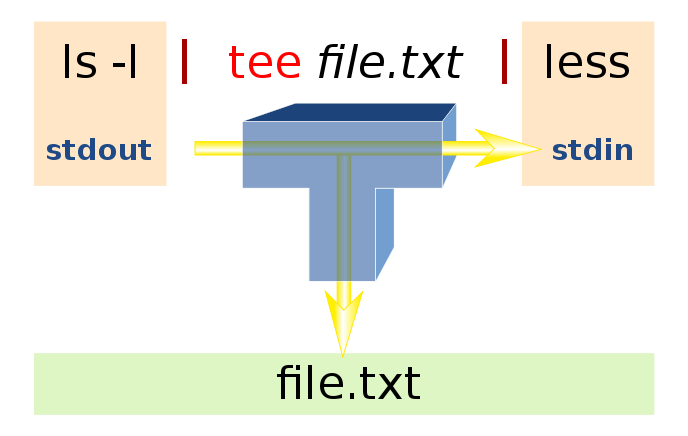
tee
- tee copies its input stream to the standard output and the files specified in argument
{kind=link}
wget
- wget is a command line utility to retrieve content from web servers.
wget http://www.uniprot.org/uniprot/ABCD1_MOUSE.fasta
- wget also supports many different options, such as ftp, authentication, etc.